
티스토리 뷰
Abstarct
DTI(Drug-Target Interaction)에 대한 Identification은 Drug discovery에서 중요한 역할을 한다.
그러나 in vitro 및 in vivo 실험으로 모든 Drug-Target pair에 대한 실험을 진행하는 것은 사실상 불가능하다.
따라서 최근 in silico computational machine learning methods들이 각광받고 많은 연구가 되고있다.
그러나 machine learning methods는 accuracy가 썩 좋지 못했고, 최근 deep learning based research가 활발히 진행중
우리는 Raw protein seq에 대해 CNN을 통해 local residue pattern을 capturing했고 다양한 길이의 subsequence에 대한 Convolution 연산을 진행했다.
CNN 연산 결과를 통해 Binding site까지 확인할 수 있었다.
Intorduction
<선행 연구들에 대한 언급>
처음 단백질의 3D structure information을 이용해 docking method를 통한 Drug-Target Interaction에 대한 simulation을 시작으로 연구되었다.
Docking method는 다양한 scoring function과 free energy를 minimization 하는 방식으로 연구가 진행되었다.
그러나 3D complex information의 부재와 simulation method의 high computational cost 문제등에 의해 docking method는 점차 주류 연구에서 밀려난다.
이후로도 다양한 방식의 simularity-based models들이 개발되었고, 그중 matrix factorization model이 가장 좋은 성능을 보였었다.
그러나 이런 simularity based model은 protein family에 대한 심한 bias가 존재해 최근에는 연구되지 않고있다.
다음으로 Feature-based model들이 연구된다. 대표적으로 Drug를 featurization하는 방식으로는 Chemical finger print가 있으며 protein의 경우 CTD descriptor라는 방식이 많이 사용되었다.
그러나 안타깝게도 Feature-based model들은 성능이 썩 좋지 못하였는데 아마도 raw한 sequence들을 featurization 하는 과정에서 잃어버리는 정보들을 제대로 복구하지 못하는데 그 이유가 있다고 생각된다.
최근에는 Deep learning based model들이 상당히 활발하게 연구되고있다.
Deep Belief Network(DBN)을 이용한 DeepDTI를 필두로하여 다양한 모델들이 만들어지고 있다.
<DeepConv-DTI 모델에 대한 간략한 설명>
우리는 특히 특정 protein family에 bias되지 않도록 다양한 protein family를 이용한 학습을 목표하였다.
Training/Valid set은 다양한 DB(DrugBank, KEGG, IUPHAR)로부터 특정 protein family에 치중되지 않도록 DTI pair를 얻었다.
Protein sequence 전반에 대해 다양한 size로 convolution연산을 진행하였다. 얻어진 Conv연산 결과에대해 global max pooling을 진행하였고 이것을 concat하여 FC layer를 지나 protein representation vector를 만들었다.
Drug sequence에 대해 RDKit을 이용해 Morgan/Circular fingerprint로 만든 뒤 다시 FC layer를 거쳐 drug representation vector를 만들었다.
만들어진 Protein/Drug representation vector를 concat하고 다시한번 FC layer를 거쳐 DTI prediction을 진행하였다.
Model의 hyperparameter optimization은 MATADOR에서 얻어진 positive DTI set과 Liu et al. 방식을 착안한 sequence smilarity based Negative DTI set sampling을 통해서 진행되었다. 다만 이 과정에서 우리의 데이터가 마치 기존의 다른 방식들보다 성능이 나쁜 것 처럼 보이는 결과를 얻는데, 이것은 positive, negative data의 수가 합쳐봐야 800개정도로 아주 작은 데이터에 대해 '파라미터 학습'을 목적으로 학습했기 때문으로 DeepConv-DTI 모델의 성능이 나빠서가 아니다. (Fig2.)
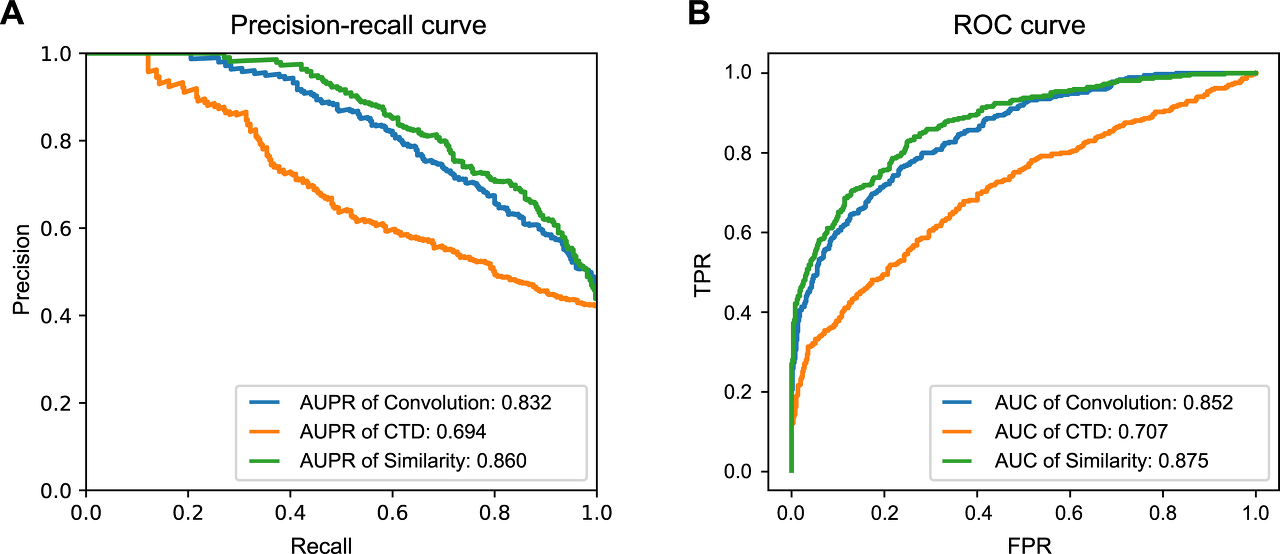
Test set으로 PubChem의 BioAssay와 Kinase SARfari의 BioAssay 데이터를 기반으로 생성한 DTI pair를 이용해 prediction task진행한 뒤 모델의 performance를 평가하였다.
Test set에 대하여 Similarity based model과 Feature based model, Deep learning based model들과의 성능을 비교평가하였고 DeepConv-DTI가 가장 좋은 성능을 보임을 확인했다. (fig4.)

Materials and Methods
<Building Dataset>
데이터는 위에서 언급한 세가지 DB로부터 얻었고, 데이터간 중복은 제거하였다. 원핵생물 및 단세포 진핵생물의 protein 데이터는 삭제하였다.
11950개의 compound와 3675개의 protein으로부터 32568개의 DTI pair를 얻었다. 그러나 DB로부터 얻은 데이터는 모두 positive이므로 negative set을 얻을 필요가 있었다. Positive data의 protein과 drug pair와 exclusive한 데이터를 이용해 random matching시켜 Negative data를 얻었다. random generation의 bias 문제를 감소시키기 위해 10 sets을 만들었다.
hyperparameter 조정은 위에서 언급한바 있으니 생략한다.
Test 데이터 생성은 크게 중요한 내용이 아닌 것 같아 생략한다.
다만 각 Train/Valid/Test dataset이 특정 protein family에 치중되지 않도록 조정하였음을 기억하면 좋을 것같다
<Drug feature representation>
Protein AA는 raw sequence를 그대로 사용한다. 그러나 Drug는 SMILES string을 RDKit을 이용해 Morgan/Circular finger print로 변형하였다. 이때 radius=2를 주었고 각각의 SMILES string은 2048 dimension의 binary vector로 input된다.

<Deep neural network model>
Protein Seq에 대해서는 CNN 및 FC layer를 이용해 latent vector로 representation 하였으며 SMILES는 언급한대로 Morgan/Circular finger print로 변형한 뒤, FC layer를 이용해 latent vector로 representation하였다. 얻어진 각각의 latent vector를 concat한 뒤, 다시 FC layer를 거쳐 output layer를 형성하였다.
Output layer를 제외한 나머지 layer에서는 actiavtion fuction으로 모두 ELU를 사용하였고, output layer에서만 sigmoid를 사용하였다.

<Convolution layer with protein embedding vector>
Protein seq를 이용한 Deep learning에서는 2가지 주요한 문제가 있다.
1. AA seq의 length가 전부 다르다. 따라서 input이 어렵다.
2. 모든 AA seq가 실제 DTI에 참여하지 않고, 특정 주요 motif 또는 domain이 주요하게 참여한다.
따라서 protein whole seq에 대한 information input은 오히려 noise가 되어 학습을 방해한다.
따라서 local residue pattern을 찾아내는 것이 중요한데, CNN은 이런 local pattern을 찾아내는데 최적화 된 모델이다.
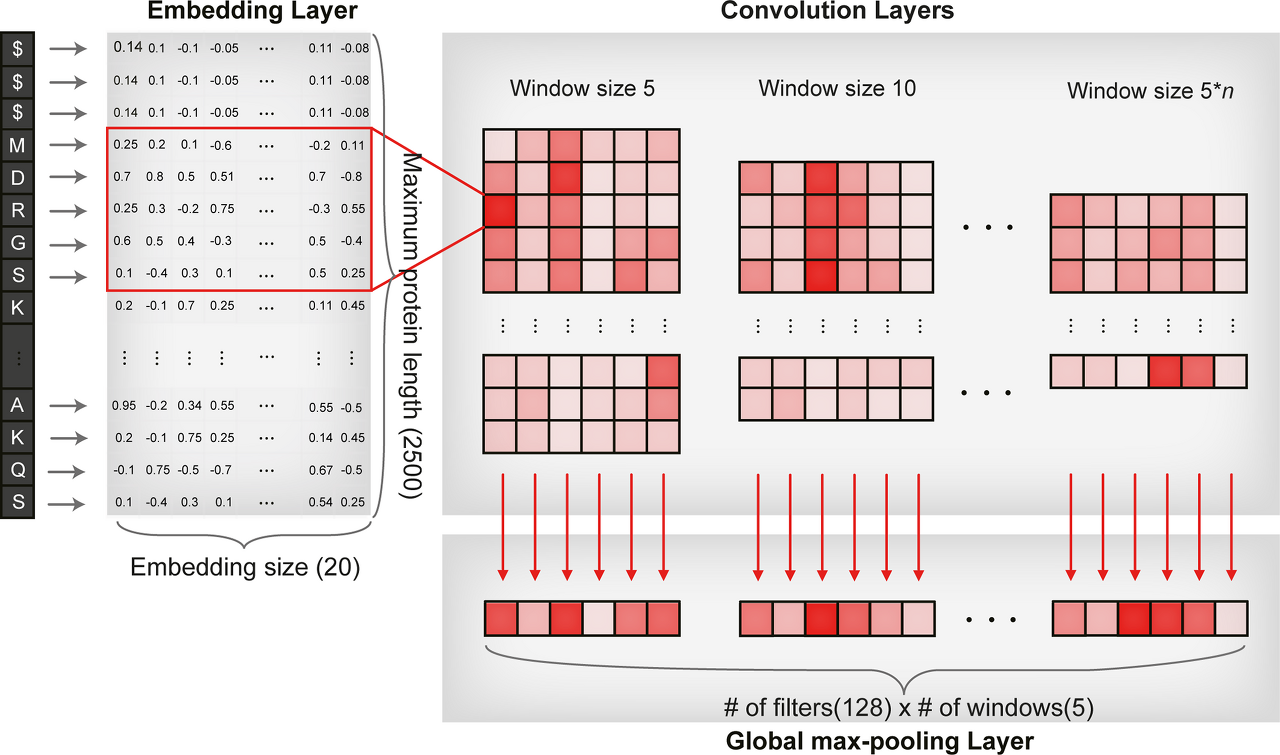
먼저 각각의 AA에 상응하는 Embedding Vector로 Transformation된다. 이 Embedding vector는 look up table로 input되는 Protein sequence의 AA를 각각의 Word Embedding Vector로 변형한다. 최초 Embedding vector는 Xavier initializer에 의해 초기화하며 이는 학습과정에서 학습 가능한 벡터이다.
1번의 문제를 해결하기 위해 Protein seq는 MAX LEN 2500으로 고정하였고 Padding/Cutting으로 length를 맞췄다. padding token으로 [$]를 사용하였으며 추후 Conv 연산과정에서 Max pooling에 의해 아무런 의미를 전달하지 못하고 drop된다.
만들어진 Protein Embedding Vector[2500, Embedding size]에 대해 1D CNN을 진행한다. Conv 연산은 stride=1, kernel_size=5*n (논문에서는 이를 Window size라고 표현한다.), filters=128인 Kernel에 의해 연산된다. 연산된 벡터의 크기는 [2500 - WindowSize +1, filters] 형태가 된다. 이것을 다시 dim=0 방향으로 global max pooling하면 [1, filters] 형태의 vector를 얻을 수 있다.
이것을 위에서 지정한 n 값에 따라 5, 10, 15, 20, 25, 30등 의 Window size별로 진행하면 총 n개의 [1, filters]형태의 vector들을 얻을 수 있다. n개의 벡터를 모두 concat한뒤, FC layer를 거쳐 protein latent vector로 representation하면 protein은 끝난다.
Drug는 위에서 언급한대로 진행하면 된다. 따로 추가되거나 설명해야할 부분이 없으므로 생략한다.
완성된 Protein/Drug latent vector를 concat한 뒤 FC Layer를 거쳐 output layer를 형성하고 sigmoid 함수를 통해 DTI prediction을 진행한다.
<Caluclation of loss and weight optimization>
Loss function은 BCELoss를 이용하며 overfitting을 방지하기 위해 L2 normalization을 추가하였다. optimizer는 Adam을 사용하였다.
<Regularization of the neural network>
overfitting 방지하기 위해 Batch Normalization과 dropout을 layer사이사이에 적용하였다.
<Analysis of convolution results>
각 window size에 대해 convolution한 결과들의 모든 filter 별 global maxpooling을 진행했기 때문에 우리는 local residue의 pattern을 highlight할수있었다. 비록 정확히 어떻게 이 max value들이 DTI prediction에 영향을 주는것인지 완벽하게 설명할 수는 없지만 더 높은 차원의 FC layer로 max 값이 전달되어 prediction 성능에 영향을 주고 있음은 알 수있다. 따라서 우리 모델이 실제로 local residue patterns를 capturing하고있다면 이렇게 높은 값을 가지는 부분은 important protein region임을 유추할 수 있다.
실제로 convolution 결과 사이사이에서 examining과 validating을 함으로써 우리 모델이 실제로 local residue pattern에 대한 capturing을 할 수 있음을 밝혔다.
sc-PDB DB는 protein과 ligand, binding site에 대한 atom-level의 complex structures 정보를 제공한다. 파싱을 통해 Vertebrata의 7179개의 protein-ligand사이의 binding site에 대한 정보를 얻었다.
얻어진 binding site와 우리가 얻은 maxpooled convolution result사이에 연관성이 있음을 가정하고 실험해보았다.
각각의 window는 128개의 pooled convolution results를 갖는다. (위에서 언급한 global max pooling 이후 [1, filters] vector를 의미) 그리고 이 pooled conv result는 global maxpooling으로부터 만들어지므로 특정 protein 영역에 대한 covering bias를 가질것으로 생각한다.
우리는 7179개의 sc-PDB entry 각각에 대해서 10000번을, randomly 128 convolution results를 생성시켰다. (? kernel size는? max pooling은? 어떻게 128 conv reuslts를 만들었다는거야?, 이부분은 약간 설명이 부족한 부분이라고 생각한다.) 그리고 만들어진 10000개의 conv results중 실제 binding site의 amino acid sequence를 포함하는 결과가 몇개나 있는지를 count하였다. 결과는 당연하겠지만 normal distribution으로 나타났다.
만들어진 normal distribution을 영가설로 설정하고 우리의 모델이 만든 각각의 window로부터 생성된 Conv result는 몇개의 binding site aa를 포함하는지 counting하고 우측 t-test 검정을 하였다.
실제로 1% 유의수준에서 1044개, 5% 유의수준에서 2172개, 10% 유의수준에서는 3019개의 sc-PDB entry에 대한 counting의 결과가 유의미한것으로 조사되었다. 따라서 우리 모델은 실제로 binding site에 대한 capturing 능력이 있다고 볼 수 있다. 이후는 visualization 내용인데 visual쪽 코드를 아직 전혀 몰라서 이해 못하겠어서 생략
<Discussion>
CNN기반 local residue pattern을 찾아낼 수 있는 novel DTI prediction model을 제안하였다. 다른 Protein descriptors보다 더 좋은 성능을 보였다. 이전의 다른 모델들보다도 좋은 성능을 보였으며, 추가적인 sc-PDB DB 데이터와의 비교를 통해 실제로 local pattern을 capturing할수있음을 보았다.
Protein중 3D structure에 대한 정보가 제공된 데이터는 많지 않다. 우리 모델은 1D AA sequence만을 가지고 진행되므로 상대적으로 많은 데이터를이용할 수 있다. 다만 우리의 모델이 좋은 성능을 보였음에도 논의해야할 부분은 있다.
첫째, 우리는 SMILES를 단순 RDKit을이용해 Morgan/Circular fingerprint로 변환하여 binary vector로 나타내었는데 drug의 추가적인 정보를 더할 수 있는 representation 방법을 생각해볼필요가 있다.
둘째, 비록 데이터가 적어 연구하기 어렵지만, 3D struture 정보를 이용하는것은 분명히 도움이 된다, 따라서 추후에는 3D structure를 이용하는 방향성에 대해서도 연구를 진행할 것이다.
'Paper > CS' 카테고리의 다른 글
| HuDiff/HuDiff-Nb 논문 리뷰 (2) Model구조 (1) | 2025.08.05 |
|---|---|
| HuDiff/HuDiff-Nb 논문 리뷰 (1) Introduction (5) | 2025.08.01 |
| Sequence-based prediction of protein binding regions and drug–target interactions(HoTS) 논문 리뷰 (0) | 2023.05.04 |
댓글
공지사항
최근에 올라온 글
최근에 달린 댓글
- Total
- Today
- Yesterday
TAG
- 파이썬
- MatrixAlgebra
- 기계학습
- MorganCircularfingerprint
- 논문리뷰
- Matrix algebra
- antigen antibody interaction prediction
- 선형대수
- 3b1b
- ai신약개발
- nanobody
- dataloader
- 백준
- 항원항체결합예측모델
- manim
- 오일석기계학습
- 인공지능
- manim library
- 이왜안
- elementry matrix
- eigenvector
- 최대우도추정
- 베이즈정리
- eigenvalue
- 3B1B따라잡기
- marginal likelihood
- Manimlibrary
- manimtutorial
- 나노바디
- MLE
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |
글 보관함