
티스토리 뷰
Sequence-based prediction of protein binding regions and drug–target interactions(HoTS) 논문 리뷰
벼랑끝과학자 2023. 5. 4. 12:58DeepConv-DTI 후속으로 나온 논문인데 figure가 조금 부실하지 않나 생각이 든다. 본문에서의 설명과 figure에서 직관적으로 이해되는 부분에서 불일치를 보이는 부분들도 있었다. (나의 이해력이 딸리는 것도 있겠지만, 썩 친절한 논문은 아닌것 같다.)
깃헙 코드를 뜯어서 이해해보자니 코드가 텐서플로우 기반이라 파이토치를 기반으로 공부한 내가 보기엔 대강 느낌은 파악되지만 명확한 이해는 안되는데다가 솔직히 코드도 조금 난잡한 것 같다.
따라서 해당 논문은 간략하게만 정리한다. 아이디어 참고만 하도록 하자.
Abstract
HoTS는 Binding Region(BR)이라는 DTI에 중요한 역할을 하는 위치를 hilights한다. BR을 찾기 위하여 Protein의 sequential motif를 찾아내는 CNN과 Drug/Protein sequential motif간의 interaction을 attention하는 Transformer를 이용한다.
HoTS는 2개의 prediction model로 구성된다. 하나는 BR predictor이고 나머지 하나는 DTI predictor이다.
3D complex DB의 data를 parsing해서 uniprot의 protein sequence로 mapping하고, 이것을 BR로 converting한다.
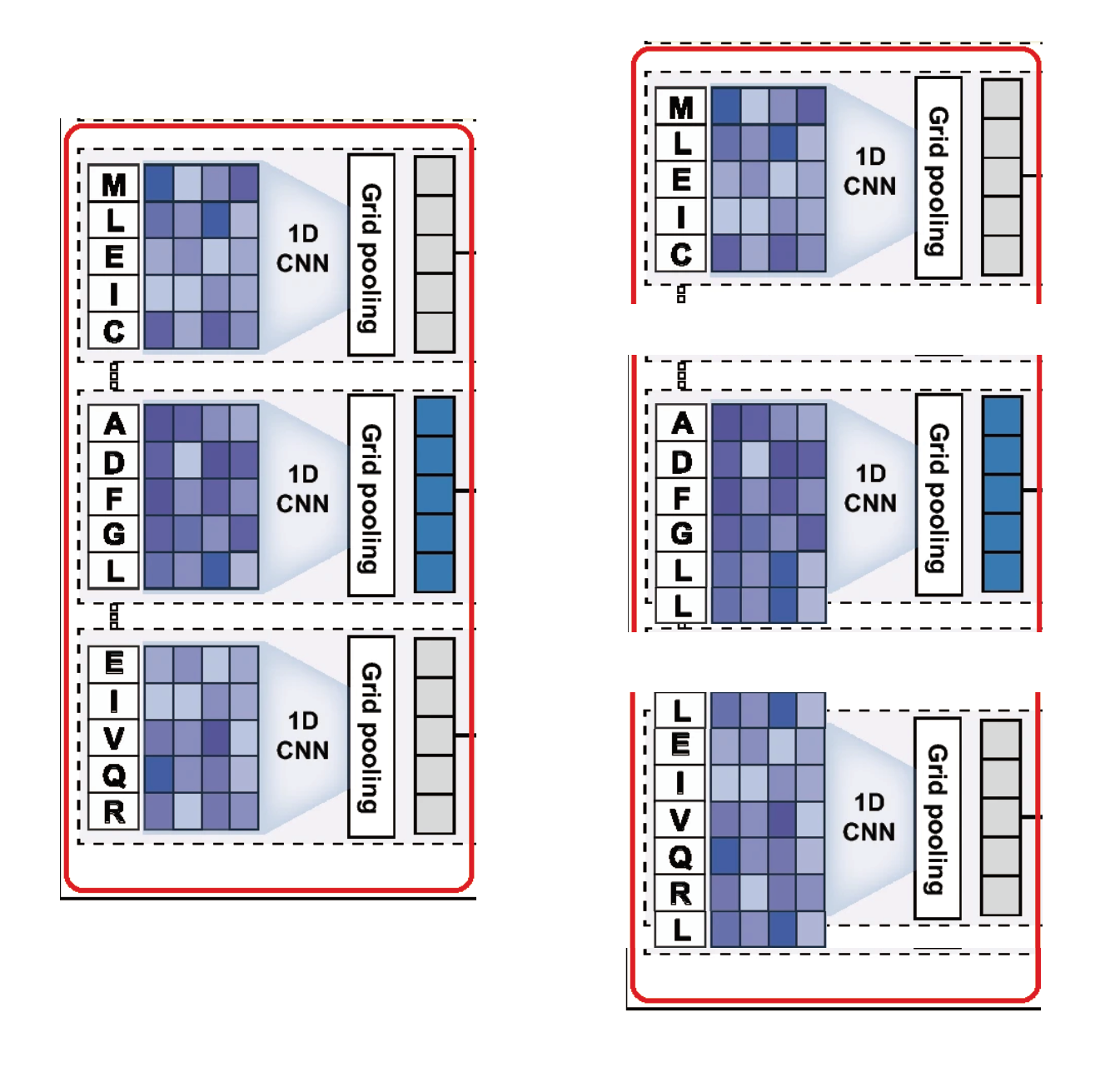
1D-CNN을 이용해 다양한 Window size 별로 protein의 motif를 extraction한다 (선행 논문인 DeepConv-DTI와 동일함)
CNN의 각 window 별로 연산된 conv result는 equal length를 가지며 각각을 protein grid라 칭한다. 각 protein grid에서 max value를 pooling하는 global max pooling방식을 이용한다.
compound는 Morgan/Circular fingerprint로 변환하여 extract된 protein grid에 concat된다.
이후 Tranformer모델을 지나면서 둘사이의 interaction을 파악한다. BR predictor를 pre-train시키면서 BR의 location, size, confidence score를 파악한다. Additional Transformer모델을 지나면서 DTI prediction에 적합하도록 compound token은 fine-tune된다.
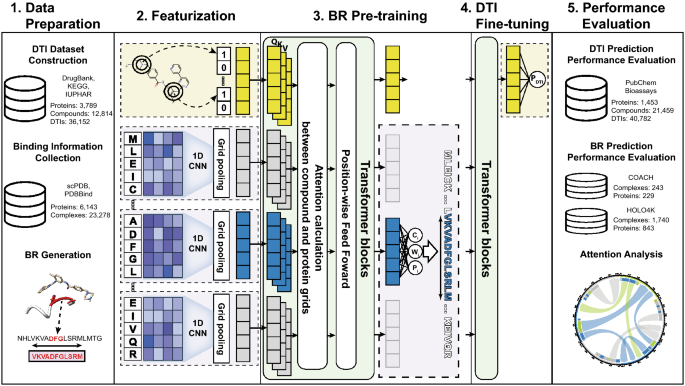
일단 내가 이해가 안가는 부분들을 정리한다.

1. 본문에는 window size별로 stride=1 sliding하며 Word Embedding vector를 Convolution 연산하는 DeepConv-DTI연산과 같은 방식을 적용했다고 한다 ( HoTS frst extracts sequential motifs, as implemented in DeepConv-DTI ) 그리고 본문 언급에도 window size별로 grid를 생성한다고 언급했었다. (A 1D-CNN with various window sizes on protein sequences then extracts motifs important to DTIs) 그러나 그림을 보면 일단 간소화를 위해서 그런것인지 Window size별로 convolution하는 내용은 완전히 생략되어있다. 오직 window size = 5일때의 figure만 넣어놓아서 혼란을 가져온다.
2. 게다가 바로 뒤에 언급되는 Concat 된 Protein/Drug representing vector의 사이즈가 H * (1 + protein_length / grid_size)라는 부분을 보면 DeepConv-DTI와는 다른 방법이 적용된 것으로 생각된다. stride=1이 아니라 stride = window_size라는 것으로 봐야하지 않나. 선행논문과 다른 grid_size라는 생소한 용어를 가지고 설명해서 또 혼란 가중, 만약 내 생각이 맞다면 본문에서 HoTS의 CNN부분은 DeepConv-DTI를 implenmentation했다고 말하고 끝낼 것이 아니라 약간 변형된 방법을 통해 진행되었다는 언급이 추가되었어야 독자들에게 혼란을 주지 않았을 것 같다.
3. 종합적으로 이야기하자면 도대체 이놈의 grid_size가 무엇을 의미하는지 파악이 안된다.
선행논문인 DeepConv-DTI를 꼼꼼히 읽고 넘어와서 읽는데도 현재 figure를 이해하는데 많은 어려움이 있는데 선행논문 없이 바로 HoTS 논문을 읽는 연구자들이 과연 해당 figure를 보면서 본문 내용을 이해하는데 명확한 도움이 될지 모르겠다.
4. 나는 개인적으로 figure에는 엄청난 디테일까진 못담더라도 굵직한 scheme들은 모두 담아야 한다는 주의인데 Grid pooling method도 너무 간략하게만 나타나있다고 생각한다. Grid pooling에 대한 부분은 다음과 같이 설명하는데 ( Next, it splits the CNN results into grids and then pools the maximum values in each grid. Te pooled maximum values are then fed into a fully connected layer. As a result, proteins are represented as sequences of grid encodings based on the convolution results, which are more suitable for predicting BRs and model interdependencies.) 과연 이 본문 내용과 figure만 가지고 grid pooling에 대한 내용을 명확히 이해할 수 있는지 모르겠다. 최소한 나는 명확한 이해는 어려웠다.
논문은 문학작품이나 시가 아니다. 독자로 하여금 '대충 이런소리라는 건가?' 하고 넘어가게 만드는 논문은 좋은 논문이 아니라고 생각한다.
5. BR Pre-training단계에서 박스친 부분에 대해서 본문에서는 Compound token과 protein grid encodings가 concat되어 Transformer로 전달되는 것 처럼 이야기하는데 (Thus, the size of the concatenated feature representing compound and sequence will be H×(1+⌈protein_length/grid_size⌉), where H denotes the size of the hidden dimension.) 그림에서는 concat되지 않은 각각의 vector들이 Tranformer로 전달되는 것 처럼 나타내었다. 전반적으로 Transformer로 input되는 vector들의 shape에 대한 설명이 부족한 것 같았다.
6. 마찬가지로 BR pretrain 이후 DTI-fine tuning 부분으로 넘어가는 부분에서 왜 protein grid의 BR 뒤에 화살표가 있는지 이해가 안간다. 무엇을 어떤 형태로 DTI Transformer Block으로 전달한다는 것인지 파악할 수 없었다.
7. BR 생성에 대해 supplementary를 붙여 더 자세히 설명해야 하지 않았을까싶다.
Methods
<BR dataset contruction>
일단 BI라는 개념이 연구마다 서로 상이해서 명확히 Binding Region에 대한 개념을 정의할 필요가 있다. 일반적인 BI 개념은 3D complex에서 ligand와 protein 사이의 거리가 threshold보다 작은 부분을 지칭한다. BI 연구에는 충분한 motif 정보의 이해가 필요한데 예를들어 3D complex에서 BI는 서로 연속적이지 않을 수 있다는 가정을 해야한다.
BI를 포함하는 새로운 구조인 BR을 제안한다. 큰 개념은 BI에 포함되는 residue를 expanding하여 BR을 만드는 것이다.
sc-PDB와 PDBBind DB로부터 BI를 parsing한뒤 uniprot Seq에 mapping 하였다. (논문에서는 해당 부분에 대한 자세한 설명들이 생략되어있는데 이 과정은 data parsing 전문 연구원들과 협업하신 것 같다.) Mapping된 Bi single Seq의 앞 뒤로 +4씩 length 9의 seq를 만들어 서로 겹치는 부분들을 merge하여 하나의 contig를 만들고 이것을 BR로 사용한다.
<DTI dataset construction>
Deep-Conv DTI와 같으므로 생략
<Proposed HoTS prediction model>
HoTS는 우선 DeepConv-DTI에서 사용한 window size based 1D-CNN을 이용해 protein의 sequential motif를 찾는다. 이후 각각의 window size에 대한 1D-CNN grid에 대하여 Maximum values를 얻어내고 pooled 된 maximum values를 FC layer로 전달한다. FC layer를 거친 값은 protein grid encodings로 변환된다.
Compounds는 RDKit을 이용해 Morgan/Circular fingerprint로 변환된다. 우선 2048개의 binary vector로 변환되어 representation된 다음 다시 FC ayer를 거쳐서 각각의 protein grid encoding vector dimension과 동일한 size로 representation한다. 이것을 compound token이라고 부를 것이다.
이후 만들어진 protein grid encoding과 compound token을 concat한다. (compound-grid feature)
compound-grid feature에 Positional Encoding을 하고 transformer block으로 input한다. TransformerBlock을 지나면서 protein과 drug는 서로에대한 interdependency, interaction 관계를 반영하게된다.
BR prediction에서는 다음과 같은 3가지 output을 prediction한다.
$$c_g, w_g, p_g = sigmoid(f_{BR}(TransformerBlocks_{BR}(h_{grid})))$$
DTI prediction에서는 다음과 같이 output을 prediction한다.
$$P_{DTI} = sigmoid(f_{DTI}(TransformerBlock_{DTI}(Transformer_{BR}(h_{compound}))))$$
그런데 위에서는 concat한 vector를 input하는것 처럼 이야기했는데 이 식에서는 h_grid와 h_compound를 따로 input해서 계산되는 것 처럼 표현하는지, 구조적으로 그게 가능한지 모르겠다.
<Prediction model of binding regions>
BR prediction은 CNN base의 YOLO와 비슷한 모델인데, 특히 3가지의 main structures로 구성된다.
1. CNN layer : protein의 seuqential motif를 찾는다.
2. Transformer : protein과 drug의 interdependency를 찾는다.
3. FC layer : 실질 BR prediction을 진행한다.
BR prediction model은 BR의 center, width, confidence score를 prediction한다. 그러나 위에서 구한 값들은 FC layer를 지나 0~1 사이의 값으로 출력되므로 이를 실질적인 유의미한 값으로 변환해주기 위해 다음과 같은 식을 이용한다.
$$Center_g = s_g + size_{grid}*c_g$$
$$Width_{ig} = r_i * e^{W_g}$$
$$FL(p_t) = -(1-p_t)^{\gamma * log(p_t)}$$
'Paper > CS' 카테고리의 다른 글
| [CVPR 2024] UniBind 리뷰 (0) | 2026.02.13 |
|---|---|
| HuDiff/HuDiff-Nb 논문 리뷰 (2) Model구조 (1) | 2025.08.05 |
| HuDiff/HuDiff-Nb 논문 리뷰 (1) Introduction (5) | 2025.08.01 |
| DeepConv-DTI 논문 리뷰 (0) | 2023.05.04 |
- Total
- Today
- Yesterday
- 오일석기계학습
- Matrix algebra
- 항원항체결합예측모델
- 베이즈정리
- Manimlibrary
- eigenvector
- eigenvalue
- 나노바디
- 3b1b
- marginal likelihood
- 선형대수
- 인공지능
- manim
- 논문리뷰
- nanobody
- manimtutorial
- 최대우도추정
- MorganCircularfingerprint
- dataloader
- 파이썬
- elementry matrix
- antigen antibody interaction prediction
- 백준
- 이왜안
- ai신약개발
- MatrixAlgebra
- 기계학습
- MLE
- manim library
- 3B1B따라잡기
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| 15 | 16 | 17 | 18 | 19 | 20 | 21 |
| 22 | 23 | 24 | 25 | 26 | 27 | 28 |
| 29 | 30 | 31 |