
티스토리 뷰
https://pubmed.ncbi.nlm.nih.gov/35293269/
Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies - PubMed
Although the therapeutic efficacy and commercial success of monoclonal antibodies (mAbs) are tremendous, the design and discovery of new candidates remain a time and cost-intensive endeavor. In this regard, progress in the generation of data describing ant
pubmed.ncbi.nlm.nih.gov
2. Learnability of AAI
본 연구자들이 정의한 Learnability of AAI prediction은 5개의 ML challenges로 나눠볼 수 있다.
- Predictability : 모델이 interaction관계를 예측하는 능력이 있어야한다.
- Generalization : A 데이터를 학습한 모델로 unseen B 데이터에 대한 어느정도의 성능이 보장되어야 한다.
- Interpretability : 해석이 가능해야 한다. (사실 나는 조금 다른 의견이다. 생물학은 해석 가능성보다는 일단 사람 목숨 살리고 보는게 우선이다. 약물로써 기능할 수 있는 후보를 찾아낼 수 있다면, 해석은 일단 뒷전의 문제라고 생각한다.)
- Model uncertainty : 동일 데이터에 대해 서로 다른 알고리즘의 모델들이 동일한 accuracy를 보일 수 있어야 한다. 왜냐면 보통 ML분야에서 training data의 rule을 완벽하게 이해하는 것은 어렵고, 다양한 모델들에서 공통적으로 학습이 가능함을 통해 dataset의 bias가 없이 학습가능한 rule이 존재함을 간접적으로 보이는 수 밖에 없기 때문이라는데... 이건 솔직히 무슨말인지 이해는 안간다
- data completeness : 데이터의 완전성, 이건 뭐 다 알다시피 Ab쪽은 데이터 확보가 참 어렵다. 좋은 데이터를 input해야 좋은 결과가 return됨은 (GIGO) 너무나 자명하다.

특히 저자들은 predictability는 sine qua non이라는 표현까지 써가면서 일단 모델이 예측자체를 못하면 뒷단은 아무짝에도 언급할 필요가 없는 필수조건임을 강조하였다. 이것은 뭐 자명하니 넘어가도록 하자.
Generalization 관점에서, 항체의 경우 similarity를 계산하는 과정에 서열만을 고려해서는 안된다는 점을 명확히 주장한다. 비슷한 서열이라도 아주 다른 target epitope를 가지는 경우가 허다하니, Ab의 similarity는 반드시 서열과 함께 binding behavior를 함께 고려해서 similarity를 결정해야 한단다. 정말 동의하는 말이지만 과연 어떻게해야 binding behavior까지 함께 고려할 수 있단 말인가? 참 간단해 보이지만 많은 생각과 연구를 해봐야 할 부분이라고 생각한다.
Interpretability는 내 관점에서는 크게 중요하지 않다고 생각한다. 간단히 chatGPT에게 해석을 의뢰하면 다음과 같은 내용이다.

지금도 영화 공공의 적을 얘기하면 떠오르는 명대사가 있다 "사람이 사람죽이는데 이유가 있나?"
나는 반대로 얘기하고싶다 "사람이 사람 살리려는데 이유가 있나?" 질병 치료제 개발에 있어서 컴퓨터 모델에서의 해석가능성은 큰 도움이 되지 않는다고 생각한다. 아니 분명 도움은 되겠지만, 그만큼의 노력을 들일 필요가 아직까지는 없다고 생각한다. 어차피 컴퓨터 모델을 분석하고 해석한 내용은 약물의 FDA 통과를 위한 근거로 전혀 채택될 수 없다. 모든 것은 어차피 직접 실험에 의해 진행되어야 한다. 아직까지 생물학은 너무나 많은 비밀들로 가득한 분야이다. 우리가 Rule based로 해석해봐야 아무런 의미가 없다.
Interpretability를 고려하기에는 우리가 아직까지 생물학 메커니즘 그 자체, 본질 자체를 이해하지 못하고 있는데 컴퓨터 계산 알고리즘에 대한 얕은 해석이 과연 어떤 의미를 가져다 준다는 것인가? 어차피 우리는 생물학적으로도 Ab와 Ag이 정확히 어떤 원리로 결합하는지 완벽하게 이해하지 못했다. 이렇게 본질조차 파악하지 못한 지식으로 컴퓨터 알고리즘의 해석 가능성이 무슨 의미가 있는가? 나는 그거 연구할 시간에 실제 치료제 개발을 위한 노력에 조금 더 시간을 쓰겠다.
(물론, interpretability 연구 자체가 의미 없다는게 아니고, 신약 개발 분야에 한정해서 아직까지 의미를 찾지 못하겠다는 말이다. CNN등의 이미지 분석에 있어서의 interpretability 연구는 필요하다고 생각한다.)
[여기서부터, 작성했던 내용이 날아가는 바람에 약간 간략하게 작성하겠다😂 ]
2.1 Formalization of antibody-antigen binding problems
DL-based로 Antibody를 연구하는 분야는 가장 크게 3가지의 목표 분야가 존재함을 언급한다.
- prediction of the AA binding interface
- prediction of Binding Affinity
- prediction of Binding partner
이건 자명하니까 여기까지만 짚고 넘어가자.
2.2 Epitope prediction
Epitope는 Antigen위에 존재하는 binding site라고 생각하면 된다. 크게 2가지 접근 연구가 존재한다. 하나는 binding partner인 Antibody(paratope)의 정보를 알고 있는상태에서, 나머지는 알지 못하는 상태에서 Epitope를 prediction하는 연구 분야 2가지이다.
이 두가지를 Antibody-Agnostic Epitope Prediction과 Antibody-Aware Epitope prediction 두가지로 언급하는데, 저자는 Antibody-Agnostic인 경우, Epitope 자체가 정의되지 않으므로 해당 분야는 biological background를 심각하게 무시한 연구로써 상당히 부정적으로 생각하고 있다. 나 역시 동의하는 바이며, 명확한 paratope 없이 Antigen에 존재하는 epitope로 작용할 수 있을법한 부위를 모두 예측하는 연구는 어불성설이고 아직까지 너무 어렵다고 생각한다.
따라서, 본 포스팅에서는 antibody-agnostic의 경우 정리하지 않고 넘어간다.
앞으로 언급되는 AAE prediction이란 용어는 Antibody-Aware Epitope prediction으로 통일한다.
2.2.1. Antibody-Agnostic Epitope prediction (생략)
2.2.2 AAE precition
이부분도 딱히 내용을 하나하나 정리할 필요는 없고, 대부분 어떤 모델이 이용되었는지를 정리해두었다. 읽는 동안 다음과 같은 모델을 언급한다.
- Bepar
- DLAB-Re
- PEASE
- PECAN
특히 볼드체 처리한 두 논문은 뒤에서 한번 더 자세히 언급한다고 하니 저자들이 보기엔 어떤 contribution이 있었나보다 싶어서 일단 다운로드 받아놓았다. 다음에 읽어보려한다.
다만, 한가지 강조하는 점은 paratope또는 antibody의 정보를 함께 사용한 (Aware) 경우의 epitope prediction 성능이 월등히 높았다는 내용을 강조하고 있다.
2.3 Paratope prediction
Antigen에 존재하는 Epitope만을 예측하지는 않는다. Antibody에 존재하는 Paratope 역시 예측이 필요하다. 저자가 말하길, 혹자들은 AAE prediction으로 예측 했으면 paratope도 자동으로 예측된거 아니냐? 라고 생각할 수 있는데 그렇지 않단다.
Epitope의 경우는 antigen이라는 거대한 단백질 덩어리 위에 존재하기 때문에 서로 sequence number끼리는 멀리 떨어진 경우라도 3D 구조상에서 가까울 수 있다. 따라서 prediction 된 AA number들이 멀리 떨어진 경우들도 있지만 paratope의 경우 대부분 CDR위에 존재하기 때문에 sequencetially close한 범위 내에서만 예측되는 경우가 대부분이라는 점과 paratope는 antibody라는 특정 mechanism을 가지고 진화해온 단백질 family인 만큼 antigen의 진화와는 전혀 다른 방향으로 구성되었기 때문에 amino acid의 선호구조 자체가 전혀 다르다는 점.
이 두가지를 이유로 paratope 역시 개별적인 prediction task가 필요하다고 주장했다.
Paratope prediction은 다음 데이터 구성 2가지 관점에서 접근한다.
- 서열 데이터와 구조 데이터
- Agnostic Epitope와 Aware Epitope
따라서 총 4가지 연구 분야가 존재하는데, 직접 연구하고 논문까지 작성해보니 경험상 Sequence based model은 25년도 시점에서 더이상 미래가 없다. 연구의 가치도 많이 떨어진다고 생각한다. 따라서 sequence based model은 생략한다. 또한 마찬가지로 저자들이 이야기했던 것 처럼 Epitope를 Angostic한 채로 Paratope를 예측하는 연구 역시 biologically reasonable하지 않다고 생각하므로 (저자들도 한번 더 이 언급을 한다) 제외한다. 따라서 나는 오직 한가지만 눈여겨서 보자 구조데이터 기반의 Antigen Aware Paratope (AAP) prediction 모델에 대해서만 정리해보자.
2.3.1 Sequence-based Antigen-Agnostic Paratope prediction
2.3.2 Structure-based Antigen-Agnostic Paratope prediction
2.3.3 Sequence-based Antigen-Aware Paratope prediction
2.3.4 Structure-based Antigen-Aware Paratope prediction
Antibody-i-Patch라는 모델은 paratope예측을 위해서 Antibody와 Antigen의 Structure를 input으로 사용한다. 추가로 ZDOCK이라는 모델을 이용했을 때 near-native poses number를 더욱 증가시킬 수 있었단다. (이게 뭔진 나중에 공부하기로 하자)
앞서 언급했던 PECAN 모델은 Paratope Epitope graph Convolution Attention Network의 약어란다. Local 정보를 graph Convolution 연산이 파악하고, Attention layer가 distant information을 파악하는데 중점을 둔 모델 구조를 갖는다. 뭐 기존의 여러 모델들을 성능적으로 제쳤다는 언급이 있으나 단점으로 가끔 너무 멀리떨어진 epitope를 예측하는 등 잘못된 예측을 하는 경우가 있었단다. 그걸 보완하기 위해 나온 모델이 Contiguous Epitope Subsampled Convolution Attention Netwokr (CE-SCAN)이란다.
ABodyBuilder라는 tool을 이용한 연구들도 많이 있다. ABodyBuilder는 서열정보로부터 Antibody 3D structure를 구조화해주는 tool이라고 보면 된다. 이렇게 ABodyBuilder로 만들어진 3D 구조의 Antibody를 ZDOCK을 이용해서 docking 시켜 paratope를 예측해보려는 시도도 있었지만, 쉽지 않았단다. 그래서 DLAB-Re라는 CNN-based 모델을 이용해서 해당 방법이 잘 동작하도록 연구가 진행된 적 있다고 한다. 정확한 모델의 구성 및 학습 방식은 논문을 직접 참고하자
Message-paasing method 기반의 Epitope-Partope Message Passing (EPMP) 모델도 언급한다. 이 모델은 Paratope 예측 모델과 Epitope 예측 모델의 combination을 통해 구성된 모델인가보다.
최근에는 Geometric Deep Learning (GDL)이라는 모델이 가장 binding interface prediction쪽에서 핫하단다. 무슨 모델인지 찾아서 읽어볼 필요가 있겠다. MaSIF라는 모델이 있다는데 읽어보자(슬쩍 찾아보니 Nature 논문이네..), 다만 이 모델은 Antibody-Antigen 보다는 Protein-Protein interaction에 치중한 모델인가보다. 따라서 Antibody-Antigen binding interface prediction 모델을 준비한다면, MaSIF 모델은 반드시 비교 모델로 넣어보는게 좋단다.
그러나 지금까지의 모델들은 대부분 molecular dynamics를 고려하지 못한다는 단점이 있다는게 가장 치명적이란다. 두 분자가 결합할 때는 분명히 sturctural(conformational) change등이 동반되기 마련이다. 이런 dynamics들이 어떤것이 있는지, 이런 것들을 더 깊게 공부하고 모델에 포함시킬 수 있다면 더욱 좋은 모델이 될 것이라는 이야기를 하였다.
2.4 From single paratope-epitope pair to many-to-many binding partner prediction
이미 알려진 epitope-paratope pair외에 binding 할 가능성이 높은 새로운 partner를 예측하는 분야도 상당히 중요하다. 저자들은 이전 본인들의 연구에서 특정 motif를 기반으로 partner를 예측하는 Deep Learning 모델을 개발했었다고 언급했다. (https://doi.org/10.1016/j.celrep.2021.108856) 심지어, 이렇게 예측된 motif들이 서로 전혀 연관 없는 경우의 Antigen-Antibody complexex에서도 shared 되는 것을 확인했고, 이를 통해 무언가 분명 antibody-antigen interaction에 중요한 interface, motif등이 존재한다는 사실을 파악했다고 한다.
저자들이 추가로 연구했던 것 중, Binding Partner를 예측하기 위한 모델에서 sequence information only의 경우는 예측성능이 상당히 저조했음을 강조한다. 데이터가 적더라도 3D 구조의 complex 데이터가 있을 때 가장 좋은 예측 성능이 나오더라는 것을 강조하였다. 매우 동의하는 바로, 앞으로 진행될 PPI 또는 AAI prediction 연구는 반드시 3D 구조의 데이터 기반이어야 한다고 생각한다.
Ab-Ligity, DLAB-VS 등 여러 모델이 있다는 언급과 함께 다양한 모델들에 대한 소개를 한다. 그런 내용에 대해서는 나중에 내가 키워드를 통해서 직접 찾아보면 될 것 같아서 여기까지만 정리하자.
2.5 Learnability of sequence-induced affinity change
Affinity 예측은 상당히 중요한 이 분야의 주요 관심사인데, 보통 다음과 같은 요인들에 의해 결정된다. ( 물론 더 있겠지만, 주요 요인들이 이렇다는 것이고)
- proximity
- contact surface area
- distribution of charged
- polar
- hydrophobic groups
DL-based 모델들은 따라서 sequence를 base로 sequential space에서 affinity space로 공간상 mapping을 하는 모델(함수)이라고 생각하면 된다.
생물학적으로 다양한 Affinity 측정방법이 있는데, 그건 뭐 넘어가고, 대부분 Free Energy of Binding (∆G, or Gibbs free energe)를 사용한단다. Structure와 complex가 모두 정해진 경우는 poteintial energy 계산방법에 의한 알고리즘 계산이 가능하단다. 보통 Docking tool 사용하면 complex간의 binding affinity가 Delta G score로 주어지긴 하는 것은 알고 있다. Guest at el.은 antibody-antigen의 docking benchmark dataset을 구성하고 20개정도 되는 affinity prediction 모델의 비교 평가를 진행했단다. 여기서 실험적으로 얻어진 실제 affininty값과 상당히 다른 결과들을 많이 보았기에, 아직까지 affinity prediction 모델도 발전해야 할 길이 멀었다는 이야기를 한다. 이 논문은 조금 참고할 필요가 있어보여서 doi 첨부한다. (https://doi.org/10.1016/j.str.2021.01.005)
그 외는 전부 Amino Acid Sequence based로 하나씩, 또는 여러개씩 mutation 또는 substitution을 해가면서 Ab-Ag 사이의 또는 Protein-Protein간의 interaction에 어떤 영향이 있는지를 연구한 모델들을 나열하는데, 이 논문을 읽으면서 주요 관심사로 볼 것은 아니라서 일단 언급된 모델들 정리만 해놓고 넘어간다.
- Computational design of antibody-affinity improvement beyond in vivo maturation (10.1038/nbt1336)
- Predicting antibody affinity changes upon mutations by combining multiple predictors(https://doi.org/10.1038/s41598-020-76369-8) 일본인논문
- mCSM-AB2 ( https://doi.org/10.1093/bioinformatics/btz779 ) 한국인논문
- mmCSM-AB( https://doi.org/10.1093/nar/gkaa389 ) 한국인논문
- GeoPPI (https://doi.org/10.1371/journal.pcbi.1009284)
2.6. High-throughput experimental methods to generate data for the prediction of antibody-antigen binding using ML
내가 서열데이터를 이용한 DTA 연구를 하면서 느낀 한계점을 저자들도 그대로 지적하고 있다. 서열데이터는 만드는데 비용이 적게 들며 매우 효율적이고 상당히 많이 만들어낼 수 있지만, Ab-Ag의 Binding에 관련된 affinity를 예측하는데 필요한 수준(resolution)의 정보를 담고있지 못한다.
최근들어 상당히 많은 Highthroughput 실험방법들로 Ab-Ag 간의 pair를 data로 만든 여러 dataset들이 등장하고 있지만, 아무래도 서열기반의 데이터로는 정말 Ab-Ag이 interaction하는지 안하는지를 알아내기에는 여전히 그 정보가 상당히 많이 부족한 수준이라서 저자는 어떤식으로든 structure 데이터를 포함한 학습데이터를 이용해야 하는 것을 강조하고 있다.
데이터 생산하는 방법론들에 대해서는 굳이 언급할 필요가 없을 것 같아서 간략하게 넘어간다.
2.7. Leveraging ground-truth synthetic data to establish lower bounds on learnability
Synthetic dataset을 추가로 구성하는 것은 필요는 하다만 simulation tool 기반으로 아주 조심스럽고 정밀하게 생성할 필요가 있고, 실제 Ab-Ag Interaction을 prediction하는데에 직접적으로 사용하기는 어렵다고 생각한단다. Novel biological inference는 실제로 experimental data를 이용할때만 얻는 것이 가능하고, Synthetic dataset은 만들어진 ML 모델들에 대한 비교평가, capacity, limitation 등을 평가하는 용도에서만 사용하는 편이 좋다고 얘기하고 있다.
이후로 뭐 본인들이 만든 Antibody-Antigen 3D structure simulation method가 있다고 설명하긴 하는데, SOTA인 AF3가 나온 마당에 굳이 다른 모델을 사용할 이유는 없어보여서 생략, 일단 Absolut!라는 모델명으로 one billion의 Ab-Ag 3D simulated strucutral dataset을 구축했다는 논문인 것 같은데, 2021년도 biobrix 등록 이후 25년도 현재까지도 publish가 되지 못한 것으로 보아 주목할만한 contribution이 없거나 리뷰과정에서 어려움을 겪고 있는 것 같다. (Target Journal이 Nature Computational Science로 IF 12.0의 상당히 레벨높은 저널이라 그럴지도 모르겠다만) 일단 알아는 두고 있자.
그외 interpretabilty와 data completeness에 대한 이야기도 하는데, 글쎄다 읽어본 바로는 그냥 일반적인 ML 관점에서의 이야기를 그냥 Ab 문제에 적용한 몇몇 관점의 이야기를 하긴 하는데.. 내가 이해가 부족한 분야라 그런지.. 그렇게 중요하지 않은 말을 너무 어렵고 장황하게 이야기하는 것 같아 보여서.. 이부분도 딱히 정리할 이유를 못느끼겠으니 넘어가겠다.
다만, 아래 Figure는 자세히 살펴 볼 필요가 있다.
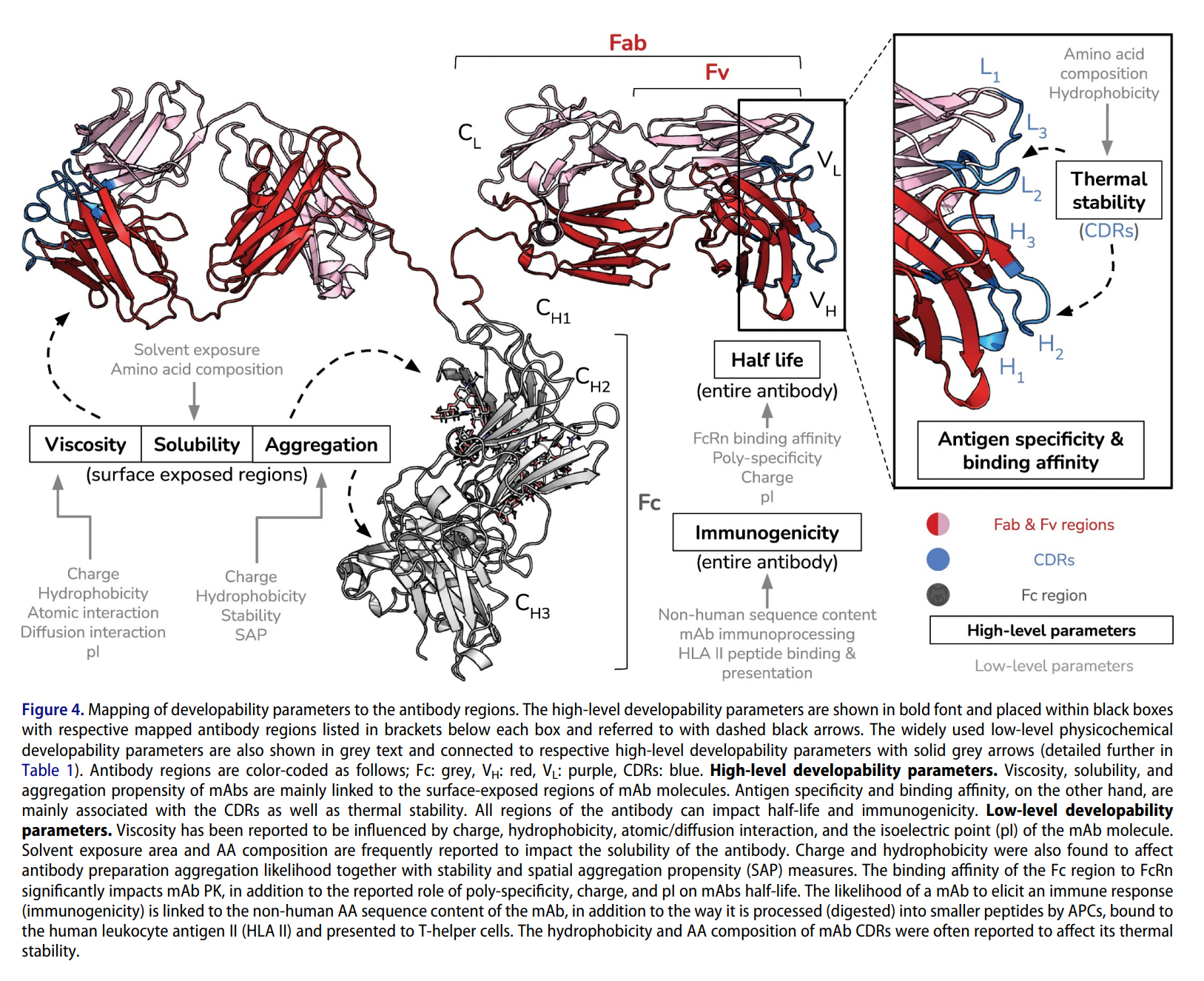
회색 글씨로 연하게 표시된 부분들은 Low-level에서 Ab design을 위해 고려해야 하는 biological feature들이고, 이것들을 기반으로 High-level의 biological feature들을 정의한 것이 검은색 굵은 영어로 나타낸 특성들이라고 한다. 나중에 모델을 이용해 Antibody를 Design할때 반드시 고려해야 할 feature들로 multi-property optimization을 사용한 생성모델을 고안해야 하니까 이 특성들은 꼭! 기억하고 참고하도록 하자
'Background > Biology' 카테고리의 다른 글
| 나노바디 (Nanobody)리뷰 (1) 구조적 특징 (0) | 2025.02.03 |
|---|---|
| Antibody Antigen Generative Modeling (3) Modularity (0) | 2025.01.31 |
| Antibody Antigen Generative Modeling (1) Introduction (0) | 2025.01.23 |
| Morgan fingerprint, Morgan circular fingerprint? (4) | 2023.04.16 |
| What are pH/Ka/pKa/Kd/Ki/IC50/EC50 values? (2) | 2023.01.11 |
- Total
- Today
- Yesterday
- nanobody
- eigenvalue
- marginal likelihood
- eigenvector
- 베이즈정리
- elementry matrix
- 3b1b
- 항원항체결합예측모델
- 파이썬
- Manimlibrary
- MLE
- manim
- 이왜안
- 3B1B따라잡기
- manimtutorial
- 최대우도추정
- antigen antibody interaction prediction
- Matrix algebra
- MorganCircularfingerprint
- 기계학습
- 나노바디
- 선형대수
- MatrixAlgebra
- 인공지능
- manim library
- 오일석기계학습
- 논문리뷰
- ai신약개발
- 백준
- dataloader
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |