
티스토리 뷰
https://pubs.acs.org/doi/10.1021/acs.jcim.5c00410
항체-단백질 복합체 데이터셋 생산에 대한 논문을 작성중이라 관련 논문을 찾아 읽어볼 필요가 있어서 정리해본다. 아마 이 논문이 우리가 진행하려던 아이디어와 상당부분 유사하기에 번역하듯 조금 꼼꼼히 읽으려한다.
1. Introduction
1. Antibody 구조설명 VL, VH, Fab, Fc, Fv, CDRs (1~3)
2. Antibody의 CDR을 computational method를 통해서 optimization 해온 역사, 특히 AI 기반이면서 structure-based model들이 가장 유망한 결과를 보임 그러나 데이터의 부재를 문제점으로 지적함
3. 현존하는 DB들의 문제점을 언급
PDB에 antibody-antigen complex들의 데이터들이 존재하고있지만 워낙 지저분하게 정리되어있어 일반화된 모델학습을 위한 적용이 어렵다는 단점을 지적한다. (그러나 이건 diffab를 보면 알 수 있지만 연구자들이 어떻게든 해결할 문제이긴 하다)
AbDb라는것은 약간 일반화되어있지만 outdated, 너무 오래되었다고 말하고 있다.
SabDab는 PDB의 모든 데이터를 모아두었고 특히 Chothia나 IMGT등의 renumbering까지 진행된 파일을 제공한다. 나름 좋은 검색 시스템을 구축하여두었지만 여전히 서로 다른 항체 타입들에 대한 formatting은 되어있지 않다는 점을 지적했다.
마지막으로 현존하는 PDB파일은 모든 Residue의 단순한 XYZ 잔기의 position만을 담고있을 뿐이고 molecule들의 물리/화학적인 description이 없다는 사실을 지적한다.
해당 논문에서 제공하는 데이터의 main contribution은 각 Residue별로 물리/화학적인 molecular description까지 제공한다는 것으로 더 많은 데이터 제공을 기반으로 더 좋은 antibody-antigen interaction을 예측하는데 도움이 되는 학습데이터를 제공할 수 있을 것을 기대함
2. Method
[요약]
파이썬 스크립트로 automation해서 RCSC PDB파일을 stnadardizing함. 근데 이때 분자간의 redocking을 이용해서 새로운 complex 결합도 in silico하게 생성하여 추가함. 여기에 마지막으로 구조의 표면과 원자 특성을 고려한 moleuclar descriptors를 추가함
2.1 Processing of Experimentally Determined Structures
[요약] 다음과 같은 순서로 RCSC PDB 파일을 standardizing하였음
1. FASTA 등록된 모든 서열 다운로드 (데이터셋 구축하는 논문이면서 과정별로 몇개씩 얻어졌고 몇개씩 드랍됐는지를 명시를 안해주는 이런 허술함은 뭐임? 어떻게된거지)
2. 단백질 서열을 항체에 맞춰 Numbering하는 ANARCI 라이브러리를 이용해서 최소 하나의 VH 또는 VL 서열이 확인된 PDB ID를 확인하여 해당 pdb파일만 다운로드, 이 과정에서 pdb 대신 cif를 제공하는 파일은 우선 cif를 받아서 pdb로 변환
3. 다음으로 데이터 필터링 진행, 하나의 조건은 아미노산 잔기에서 atom 정보가 없는 것 들을 제거하였고 일반적이지 않은 구조의 데이터 (예를들면 double variable domains라는데 이게 뭘 의미하는건지 설명해줘야하지 않나? supplementary에라도 어떤 데이터들이 누락된건지 자세히 설명해줘야한다고 생각함) 제거하였음
4. 결과적으로 최종 파일들은 하나의 파일에 하나의 complex만 담겨있는 standardizing된 파일들만 남았음
5. 이걸 2가지 서브셋으로 다시 나누는데 하나는 antigen이 없는 antibody 구조만 들어있는 파일들과 antigen과 함께 들어있는 complex 파일들로 나눔. 단, 이때 antigen의 길이는 50AA 이상인 것들만 선택함
[디테일]
Step 1 : Renumbering the residues and renaming the antibody chains
항체의 chain들을 파악하고 그들의 AA residue를 확인 한 뒤, ANARCI의 Martin scheme을 이용해서 renumbering하였다. 이 과정에서 HC와 LC를 지정한 이니셜은 모두 H, L로 변경하였고 만약 scFv라면 HC과 LC를 h, l로 변경하였음.
항원의 경우에는 pdb파일에 등록된 이니셜을 그대로 사용하되, 만약 그 이니셜이 H나 L로 지정되어있으면 그것은 일괄로 'A'로 변경함
Step 2 : Identification of VH-VL pairs and antigens with biological interfaces
pdb에 주어는 antibody가 실제로 하나의 쌍으로 존재하는 온전한 항체 서열인지를 확인하기 위해서 Martin renumbering된 서열 기준 VH의 Cys92와 VL의 Cys88 사이의 Carbon Alpha 거리가 22Å 이하인것만 선별.
다음으로 만약 pdb파일 내에 antigen chain들이 여러개 들어있으면 PRODIGY-CRIST를 이용해서 실제 biologically 의미있는 구조인지를 파악하였음. 만약 PRODIGY-CRIST 검사결과 biological interface가 맞으면 oligomeric chains로 판단하였고 그렇지 않은 경우는 X-Ray 구조결정 실험의 필요조건인 고농축 과정에 의한 crystallographic interfaces(비 생물학적인 환경에 의한 강제접촉면)으로 판단하여 제거하고 monomer로 사용하였다고 함.
(이 부분은 detail한 설명이 더 필요할 것 같음, PDB파일과 X-Ray 구조화 실험에 대한 지식이 없으면 이해하기 어렵고 또한 oligomer, monomer로 사용한다는게 원본은 어떻게 생겼는데 어떻게 수정했다는 것인지 논문을 보고는 이해가 명확하게 될 수가 없음, 다시 말하지만 데이터 구축을 main contribution으로 하는 논문이라면 supplementary를 적극 활용해서 데이터 구축 과정을 명확하게 설명해야하는데 그런게 없이 논문이 된게 참 아이러니함)
Step 3 : Construction of antibody-antigen complexes
monomer는 상관없는데 oligomeric antigen chain을 가지는 pdb파일은 각각의 complex로 나눠야 함. 항체의 CDR Residue와의 거리가 7.5Å 이하인 것이 하나라도 존재하면 해당 항체와 그 항원의 Chain만 따로 parsing해서 새로운 pdb파일로 저장하며, 기존 pdb_id를 기반으로 XXXX_n.pdb로 저장함. (이미지 참고)
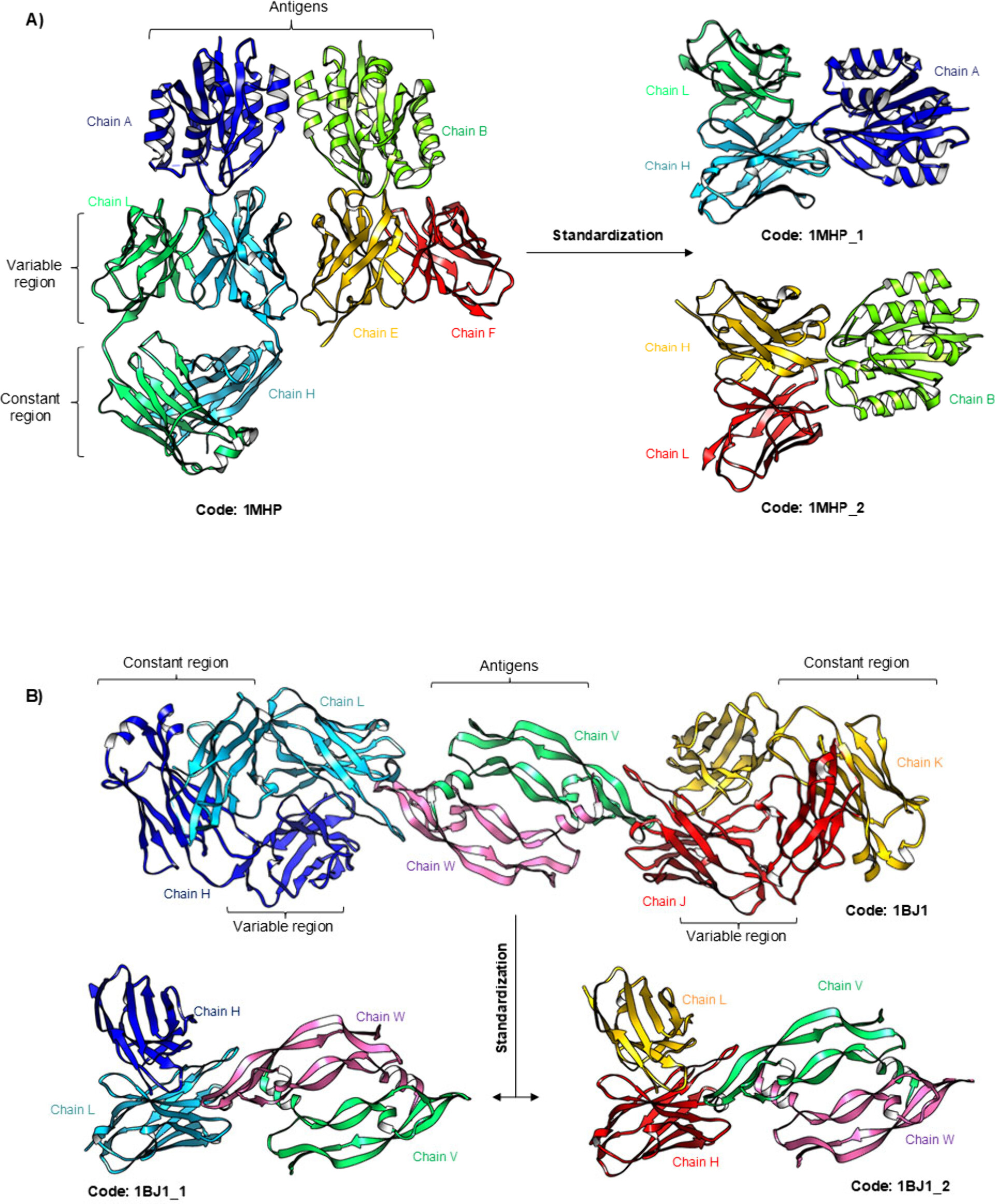
Step 1~3까지 거친 파일들을 이용해서 AbSet이 구축되었으며 statistical profile들이 계산되었고, 그 외 resolution이나 method등의 descriptor들을 PDB에서 추가로 파싱하여 저장
2.2 Construction of the In Silico Subset
Ag-Ab complex의 데이터 증강을 위해서 in silico method를 사용하였으며 2가지의 메인 전략을 사용하였다.
Docking으로 복합체를 인위적으로 더 생성 → 1. PDB 체인 그대로 사용 2. 항체 구조를 모델링한 뒤 Docking
→ 올바른/잘못된/근접한 결합 모드를 모두 포함시켜 항체–항원 상호작용의 변동성을 반영.
2.2.1. Molecular Modeling
항체 모델링은 FASTA기반으로 AbBuilder2를 이용했음 이 논문은 읽어보길 추천함 (해당 부분에서 내 논문에 필요한 argument : 나는 지금 AF3기반으로 항체의 구조를 모델링하고있음. 항체 한정 AF2 기반의 모델링보다 AbBuilder2가 유사하거나 더 좋은 성능을 보일 가능성이 높음. 그러나 AF3는 아주 최신으로 더 업데이트 되어서 나온 논문이기 때문에 성능이 더 향상되었을 가능성이 높음. 직접적인 비교실험으로 보일 필요 있음)
예측된 구조에 대한 성능은 원본의 CDR 구조와 예측된 CDR의 RMSD를 이용하는 듯
2.2.2. Molecular Docking
Redocking은 binding pose가 잘 정립되어있는 경우에 한정하여 두 molecule들의 interaction을 analyzing하는 연구이며 Haddock 2.4를 이용해서 진행되었다. HDock이 필요로하는 조건에 맞춰 Residue의 overlapping이 없도록 HC 1~500, LC 501~로 residue number를 재지정하였다. 또한 Docking하기 위해 필요한 active residue들은 항체의 CDR에 위치한 부분들을 지정하였다.
실험적으로 증명되어 PDB가 명확히 존재하는 데이터에 대해서는 blind approach가 사용되었는데 항원의 어느부분이 epitope인지 명확히 알 수 없으므로 active residue 주변의 Residue들을 한정하여 표면에 노출된 모든 잔기를 passive residue로 처리하였다.
FreeSAS C library를 이용해서 Solvent-Accessible Surface Area(SASA) 값을 계산하고 이 값의 변동을 이용해서 Pseudo-epitope를 Active Residue로 지정함
대충 두 가지 서브셋의 구성을 표로 정리하면 다음과 같음
전략 1: 실험 구조 기반 (Blind Docking)
PDB → CDR active + 항원 전체 passive → blind docking
전략 2: 모델링 기반 (Non-blind Docking)
서열 → AbBuilder2 → HDock blind docking → FreeSAS 기반 epitope active 지정 → HADDOCK refinement

HDock의 MolProbity를 이용해서 지정해줘야하는 히스티딘의 성질을 설정해주고 Docking을 실행함
이때 HDock은 하나의 complex에 대해 docking을 실행하면 다음 세 가지 스텝별로 pdb 결과를 반환해주는데
- randomization of orientations and rigid body minimization (it0)
- semiflexible refinement (it1) of the torsion angles ϕ, ψ, and ω
- explicit solvent refinement (itW)
논문에서는 하나의 complex에 대해서 it0, it1, itW 별로 각각 1000, 250, 250개씩의 데이터를 생산했다고 함.
2.2.3. Evaluation of Docking Results
HDock에 의해서 계산된 docking pose는 다양한 물성을 고려하여 DockQ라는 하나의 quality 점수로 환산되는데 이때 점수의 범위에 따라 4가지의 범주 High / Medium / Acceptable / Incorrect로 나눴다고 함.
그런데 이 부분은 아주 치명적인 에러를 가지는데 DockQ는 원래 native complex(원본 실험 구조)가 있어야 계산 가능한 지표임(Fnat, LRMS, iRMS 모두 원본과 비교 필요함). 그런데 이 논문의 전략 2 (AbBuilder2 모델링 + HDock)는 native 구조가 없으므로 사실상 pseudo-native (예: HDock top pose)를 정답처럼 취급했을 가능성이 큼. 따라서 여기서 계산되는 DockQ를 절대적 품질 지표로 쓰긴 어렵고 단순히 상대적 분류용 보조 지표 정도로 봐야 하는데 그런 limitation 언급 없이 마치 절대적인 평가지표로 사용 가능한 것 처럼 축소해서 말해두었음. 이것은 분명한 오류라고 보여지며 반드시 한계점으로 명확히 언급이 되었어야 할 부분이라고 생각됨.
2.3. Molecular Descriptors Calculation.
다음과 같은 descriptor들을 추가하였다. (이것도 이해가 안감. 무슨 tool을 이용해서 어떻게 얻었는지, 데이터에 어떻게 추가되어 사용할 수 있는지 활용성에 대한 내용 등을 supplementary에라도 추가해서 설명해야하는데 그냥 descriptor를 추가로 얻어서 제공한다고 한단락에 적어두고는 그냥 넘어가는건 데이터를 제공하는것이 main contribution인 논문에서는 용납되기 어려운 무책임한 서술같은데... 이게 어떻게 리뷰어들에게 아규가 안되고 논문이 되었는지 참 신기할따름)
- Solvent Accessible Surface Area
- Relative Accessible Surface Area
- Atomic depth
- Potrusion index
- Hydrophobicity
- Sequence
- Half-sphere exposure calculations
- Cα coordinates
- ϕ and ψ dihedral angles
- Secondary structure of the protein
3. Results
3.1. Identification and Standardization of Antibodies.
PDB의 FASTA를 기반으로 얻어진 데이터는 최종적으로 9169개의 antibody entries가 남았다. (AbSet)
반면 SabDab는 2024년 12월 20일 기준으로 9026개의 antibody entries만 등록되어있었다.
왜이런지 분석해야했고, AbSet과 SabDab를 비교했더니 서로 일치하지 않는 부분들이 있었고, 더 자세한 조사를 통해서 완전한 교집합을 보이는 부분 데이터셋만을 남겨 최종적으로 7449개의 항체 구조만 남았다. (자세한 내용은 생략)
다음, oligomeric 항원 데이터들을 정리하는 standardizing 과정을 거치면서 XXXX_n등으로 데이터가 늘어나므로, 최종적으로 표준화 과정을 거쳐서 14184개의 PDB파일로 정리되었다.
항체 구조 분포
- HC_LC 쌍 10495개,
- 단일 HC 2852개,
- 단일 LC 622개,
- scFv 215개로
항원 구조 분포
- 단백질 9146개
- 항원이 없는 자유항체 구조 5038갸
3.1.1. Crystallographic Resolution and Antigen Diversity of AbSet.
분자 구조 데이터베이스를 분석한 결과
- 분석 방법: 구조의 대부분(72%)은 X선 결정학으로, 나머지는 주로 전자현미경으로 구성됨
- 데이터 품질: X선 결정학으로 얻은 구조의 정밀도(해상도)가 더 높았고, 전체 데이터의 86%가 신뢰할 만한 수준 (4Å 이하)
- 핵심 연구 대상: 구조 중 21%가 코로나19 바이러스의 스파이크 단백질과 항체의 결합에 관한 것으로, 팬데믹 기간 동안 이 분야의 연구가 집중되었음을 보여줌
3.2. In Silico Data set.
in silico generated 데이터셋은 우선 1755개의 항체들이 선정되었고, 그들의 FASTA를 추출하여서 AbBuilder2를 이용해서 pdb파일을 구성하고 HDock을 이용해서 위치-지정기반 docking을 수행함. (Method에서 설명한 전략2)
추가적으로 2135개의 실험기반 구조체가 결정된 항체들을 이용해서 blind docking을 통해서 데이터를 추가로 구성하였다. (단순 미세조정수준의 결과, 전략1)
3.2.1. Antibody Modeling.
AbBuilder2를 이용했고 몇몇 outliers들을 제외하고는 대부분의 CDR에서 0.3 수준의 차이를 보였다.
(누누히 말하고 있지만, 이건 다시말하면 아주 미세하게 변형된 수준의 데이터들이 중복되어서 만들어지고 있다는 것을 의미함. 항원-항체 데이터의 부족함을 보완하기 위해 제공되는 데이터임에도 불구하고 큰 변화 없이 단순한 미세조정된 수준의 중복 데이터를 제공하는 것이 학습에 어떤 의미를 가질 수 있는지 의문임)
3.2.2. Molecular Docking.
전략 1과 전략 2를 사용해서 각각 438750개씩 데이터를 생성했고 DockQ score에 따라서 4개의 클래스로 분류.
(데이터 논문인데 데이터 클래스 분포 숫자에 에러를 내놨음 ㅋㅋ)
이 데이터는 우리가 만들고자하는 데이터와는 성질이 완전히 다름. 이 데이터는 오히려 DockQ 값이 매우 낮은 class의 데이터가 대부분이고 학습할 수 있는 정답값의 정보가 두 단백질의 interaction이 단순한 DockQ 스코어를 기반으로 설정된 값인데 학습모델이 뭘 학습할 수 있다는 것인지가 의문임... 현재 만드는 데이터는 이렇게 '의미없는' 데이터가 되지 않도록 조심해야겠음
3.3. Implementation of Molecular Descriptors.
논문 서술이 허술해서 여기까지만 읽고 나머지는 그냥 AI로 정리... 더이상 읽어볼 필요는 없어보임
- 데이터 특징 추출:
- 실제 구조: PDB 데이터베이스에서 얻은 모든 실제 항체 구조에 대해 '분자 기술자'(분자의 특징을 나타내는 숫자 값)를 계산했습니다.
- 가상 구조: 컴퓨터 시뮬레이션으로 만든 가상 구조들은 전부 계산하지 않고, 각 시스템당 250개 중 **대표 4개(고품질, 중간, 수용 가능, 부정확)**만 뽑아서 계산했습니다. 만약 좋은 품질의 구조가 없으면 '부정확' 구조로 4개를 채웠습니다.
- 데이터 저장: 계산된 모든 특징 값은 CSV 파일(엑셀과 유사한 형태)로 저장했습니다.
- 성능 테스트:
- 효율성 검증: 특징을 계산하는 프로그램이 얼마나 빠른지 16개 구조를 대상으로 테스트했습니다.
- 결과: 계산 시간은 데이터 양에 비례하여 예측 가능하게 증가했으며(파일 1개에 10초, 전체 테스트에 40초), 6코어 프로세서에서 안정적으로 작동했습니다.
- 결론: 계산 과정이 빠르고 효율적이어서 메모리 부족 같은 문제는 발생하지 않았습니다.
한줄평 : 아니 미친 어떻게 데이터 배포를 main contribution으로 쓴 논문이 데이터 활용성에 대한 검증결과가 하나도 없음? JCIM 나름 나쁘지 않은 저널인데... 이 논문은 솔직히 운이 조금 좋았다고 생각됨.. 아마 리뷰어들이 관련분야 전문가가 전혀 없었던 듯
'Paper > Bioinformatics' 카테고리의 다른 글
| DeepInterAware 논문 리뷰 (2) 실험 및 결과 (4) | 2025.08.26 |
|---|---|
| DeepInterAware 논문 리뷰 (1) 모델 구조 (1) | 2025.08.25 |
| AttABseq 논문 리뷰 (4) | 2025.08.22 |
| [2022 NIPS] DIffAb 논문리뷰 (1) (0) | 2025.03.27 |
| HyperAttentionDTI 논문 리뷰 (4) | 2023.11.24 |
- Total
- Today
- Yesterday
- 이왜안
- manim
- MatrixAlgebra
- 최대우도추정
- 백준
- MLE
- marginal likelihood
- 파이썬
- manimtutorial
- MorganCircularfingerprint
- Matrix algebra
- 기계학습
- Manimlibrary
- 선형대수
- 베이즈정리
- nanobody
- 인공지능
- ai신약개발
- 오일석기계학습
- 항원항체결합예측모델
- antigen antibody interaction prediction
- 3B1B따라잡기
- eigenvalue
- 논문리뷰
- eigenvector
- 3b1b
- manim library
- 나노바디
- elementry matrix
- dataloader
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |