
티스토리 뷰
https://biomadscientist.tistory.com/185
DeepInterAware 논문 리뷰 (1) 모델 구조
https://pmc.ncbi.nlm.nih.gov/articles/PMC11967782/ DeepInterAware: Deep Interaction Interface‐Aware Network for Improving Antigen‐Antibody Interaction Prediction from SequenceAbstract Identifying interactions between candidate antibodies and target ant
biomadscientist.tistory.com
이 논문 배울점이 많다. 모델 설명도 구체적이고 아이디어도 뭐 엄청나게 신박하지는 않지만 깔게 마땅히 없이 직관적으로 잘 만들어두었다. 거기에 본 포스팅에서 다루겠지만 실험 설계도 아주 체계적이고 다양하다. 검증 단계가 풍부하고 잘못되지만 않았다면 리뷰어들도 사람인지라 쉽게쉽게 넘어갈 수 있다. 본 연구에서 제안한 검증 방식들과 검증에 사용된 데이터들은 꼭 응용해보면 좋겠다.
크게 4가지 검증이 존재한다. 하나씩 살펴보자
- Ag-Ab binding datasets
- Ag-Ab neutralization datasets
- Binding Site Identifications
- Binding free energy change dataset
사용된 데이터셋을 일단 전반적으로 GPT를 이용해서 요약해 보았으니 참고하자.
🔹 전체 해석
1. AVIDa-hIL6
본 연구에서는 DeepInterAware의 성능을 평가하고 결과를 뒷받침하기 위해 여러 데이터셋을 사용하였다.
구체적으로 AVIDa-hIL6와 SAbDab 데이터셋은 결합(binding) 예측에 사용되었으며, HIV와 CoV-AbDab 데이터셋은 중화(neutralization) 예측에 활용되었다. 또한 SAbDab 데이터셋은 결합 부위(binding site) 예측에도 사용되었으며, AB-Bind와 SKEMPI2 데이터셋은 결합 자유에너지 변화(ΔΔG) 예측에 활용되었다.
- AVIDa-hIL6: 항체의 variable domain(VHH)에 대한 항원–항체 상호작용(AAI) 예측용 포괄적 시퀀스 데이터셋.
- 항원은 wild-type IL-6 단백질과 30개의 변이체.
- 총 20,980개의 binding pair, 552,911개의 non-binding pair.
- 연구에서는 ANARCI로 항체의 CDR loop를 추출하고, 동일 CDR loop를 가진 중복 쌍을 제거 → 최종 10,178 binding, 315,708 non-binding.
- 실험 설정: 다섯 번의 독립적 실험(랜덤 시드 다르게), 각 실험에서 15개의 항원 변이체를 테스트셋으로 두고, 나머지는 9:1 비율로 train/validation. 결과의 평균과 분산을 보고.
2. SAbDab
- SAbDab: Protein Data Bank(PDB)에서 수집된 모든 항체–항원 복합체의 포괄적 데이터베이스.
- 항원 서열이 50 아미노산 이상인 경우 선택 → 1,193개 복합체.
- 동일 항체 CDR loop 기준 중복 제거 → 최종 1,513 Ag-Ab pair.
- Huang et al. 방법에 따라 CD-HIT로 항원/항체 서열 상동성 계산. 항원 서열 상동성 0.9 기준으로 772 subgroup 생성.
- 동일 subgroup 안 항체–항원은 결합 가능성이 높음, 다른 subgroup 간은 결합 확률 낮음.
- 최종적으로 binding pair 2,131개, non-binding pair 2,131개.
- 추가로 ClustalW를 이용해 항원 subgroup들로 계통수를 만들고, 772개 subgroup을 7개 cluster로 분류.
- 실험 설정:
- Binding prediction → 각 cluster에서 4:1 비율로 train/test split, 다섯 번 독립적 실험 후 평균/분산 보고.
- Binding site prediction → 1,513쌍에 대해 5-fold cross-validation, 평균/분산 보고.
3. HIV
- HIV dataset: Human Immunodeficiency Virus 관련 항체–항원 중화 데이터.
- Zhang et al. protocol 따라 항원과 항체 모두 상동성 0.9 이상인 쌍 제거.
- 최종적으로 neutralization pair 24,907개, non-neutralization pair 26,480개, 항원 1,752종, 항체 457종.
- 실험 설정: 3가지 시나리오 설정
- Ab Unseen: 항체 중 15%를 unseen으로 두고 해당 pair는 테스트셋. 나머지 9:1로 train/valid.
- Ag Unseen: 항원 중 20%를 unseen으로 두고 해당 pair는 테스트셋. 나머지 9:1로 train/valid.
- Ag & Ab Unseen: Ag Unseen 기반, 추가로 test에 등장한 항체 pair를 train에서 제거.
- 각 시나리오마다 다섯 번 실험 후 평균/분산 보고.
4. CoV-AbDab
- CoV-AbDab: 다양한 코로나바이러스에 결합 가능한 conventional antibody 및 nanobody 정보 DB.
- 항체 서열과 함께 neutralization/non-neutralization 관계 수집.
- 단, CoV-AbDab에는 항원 이름만 있고 서열은 없음 → 레퍼런스에서 receptor-binding domain(RBD) 서열을 확보 후 antigen으로 사용.
- 중복 pair 제거.
- 최종적으로 항원 30종, 항체 4,235종, neutralization pair 5,486개, non-neutralization pair 9,110개.
- 실험 설정: transferability 실험, 다섯 번 실험 수행.
- 각 split에서 30%는 transfer train set, 70%는 test set.
- HIV와 동일하게 상동성 0.9 이상은 제거.
- 평균/분산 보고.
5. AB-Bind & SKEMPI2
- AB-Bind: 32개의 항체–항원 복합체에서 1,101개의 변이에 대해 실험적으로 측정된 binding free energy 변화(ΔΔG).
- light chain & heavy chain annotation이 있는 경우만 사용 → 654 mutant.
- SKEMPI2: 단백질–단백질 상호작용에서 변이에 따른 결합 에너지, 동역학, 열역학 변화 데이터.
- 역시 항체–항원 mutant만 추출 → 1,021 mutant.
- ΔG는 해리상수(Kd)로부터 ΔG=RTlnKd\Delta G = RT \ln K_d로 계산 (R = 1.987e-3 kcal·mol⁻¹·K⁻¹, T = 298K).
- 실험 설정: binding free energy change prediction → 두 데이터셋 모두 10-fold cross-validation, 평균/분산 보고.
🔹 데이터셋별 요약표
| AVIDa-hIL6 | Binding prediction | IL-6 WT + 30 mutant, 최종 10,178 binding / 315,708 non-binding |
| SAbDab | Binding prediction & site | PDB 기반, 최종 1,513 pair, binding 2,131 / non-binding 2,131 |
| HIV | Neutralization prediction | 24,907 neutralization / 26,480 non-neutralization |
| CoV-AbDab | Neutralization prediction | 항원 30, 항체 4,235, 총 14,596 pair |
| AB-Bind | ΔΔG prediction | 654 mutant |
| SKEMPI2 | ΔΔG prediction | 1,021 mutant |
2.2. DeepInterAware Outperforms the State-of-the-Art Methods in Antigen–Antibody Interaction(AAI) Predction
여기에서는 AAI prediction과 Antigen-Antibody Neutralization(AAN) prediction에 대한 검증을 진행했음. 우선 AAI는 AVIDa-hIL6와 SAbDab데이터셋을 사용했고 Neutralization 검증에 대해서는 HIV 데이터셋을 사용했음.
여기에 추가로 HIV와 함께 CoV-AbDab dataset을 사용해서 모델이 다른 Antigen에 대한 평가도 진행했다. 우선 AAI prediction 성능 비교표를 보면 다음과 같다.
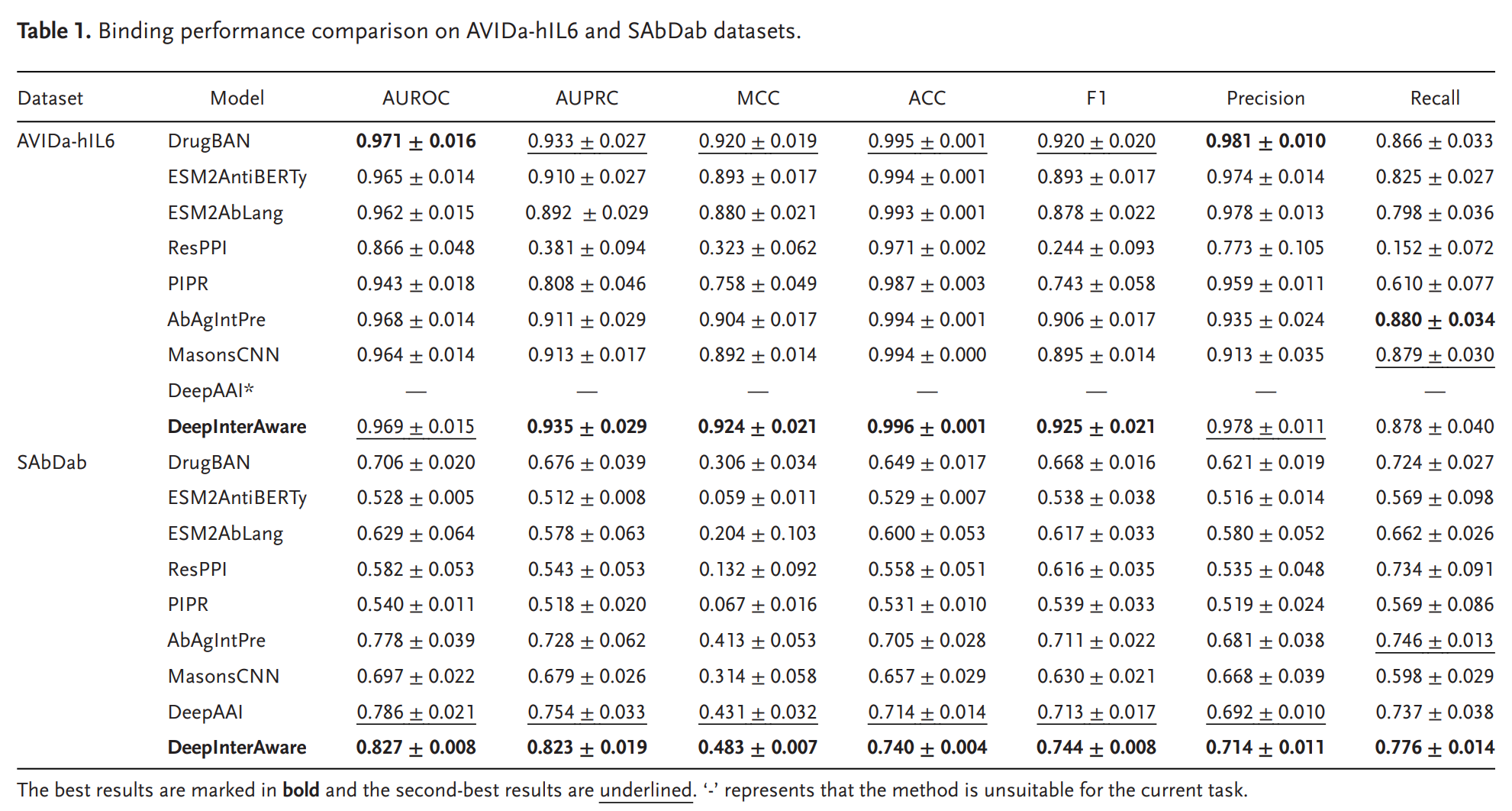
대개 그렇지만 본 연구의 논문이 가장 좋은 성능을 달성했다. AVIDa-hIL6 데이터셋보다 SAbDab 데이터셋이 더 어려운 데이터라서 예측 성능은 전반적으로 하락하지만 다른 비교모델에 비해서 더 좋은 성능은 항상 유지했다.
또 항체가 항원을 무력화하는 가장 주된 방법이 중화(Neutralization)인데 해당 항체가 그 항원을 중화시킬 수 있는 능력이 있는지를 예측하는 AAN 성능에 대한 비교 표도 존재한다.
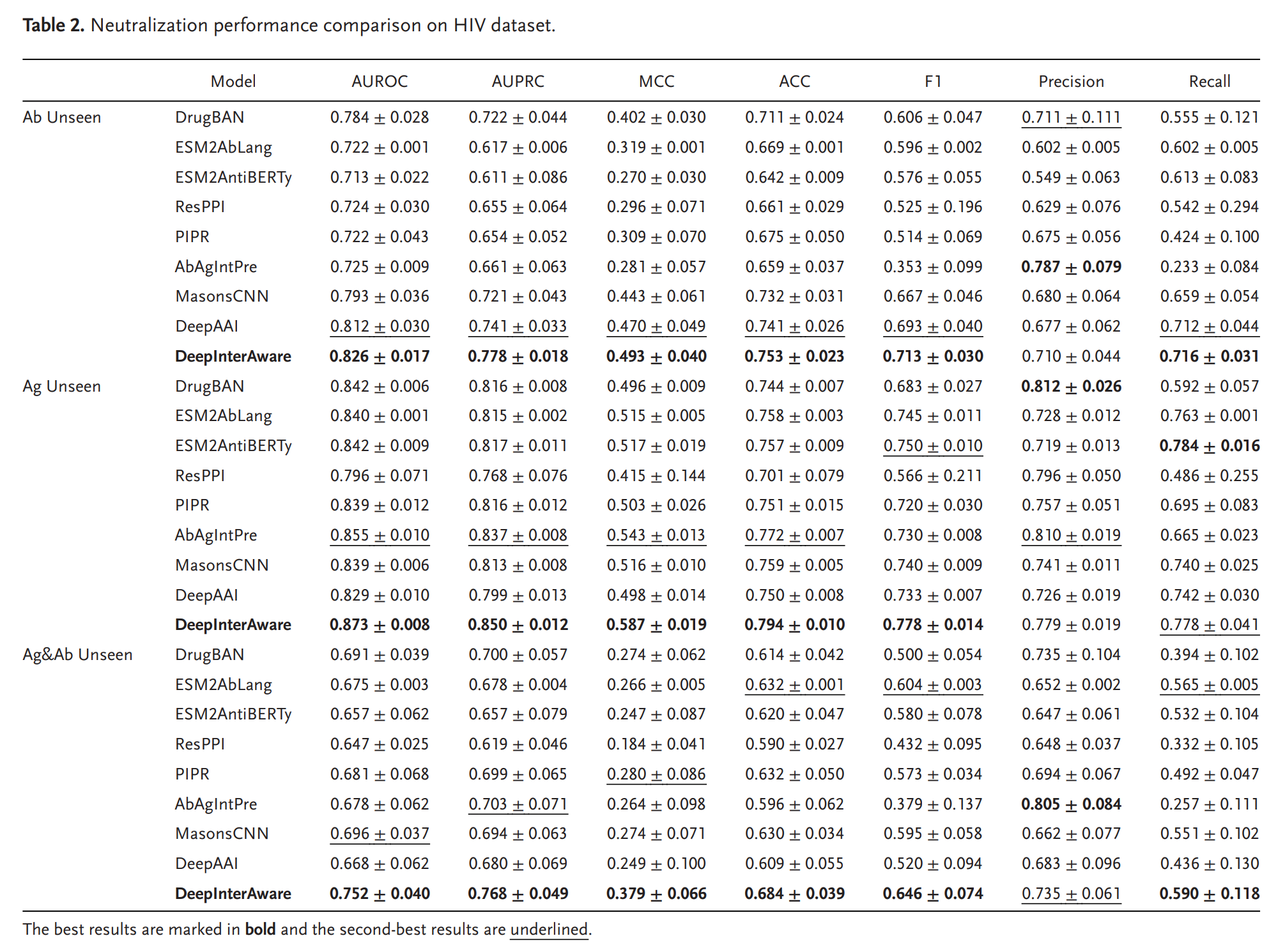
특히 항원 항체 모두에 대해서 Unseen인 경우의 성능 차이가 크다는 점에서 강조하였다.
다음으로는 모델이 다른 antigen에 대해서도 예측 성능을 유지를 잘 하는지(transfer), 학습된 특성을 다른 항원에 대해서도 잘 적용하는지를 보기위해 먼저 HIV 데이터셋으로 모델을 pre-training한 뒤, CoV-AbDab 데이터로 fine-tuning 한 뒤, CoV-AbDab데이터로 테스트를 진행한 Transferability performance 결과이다.
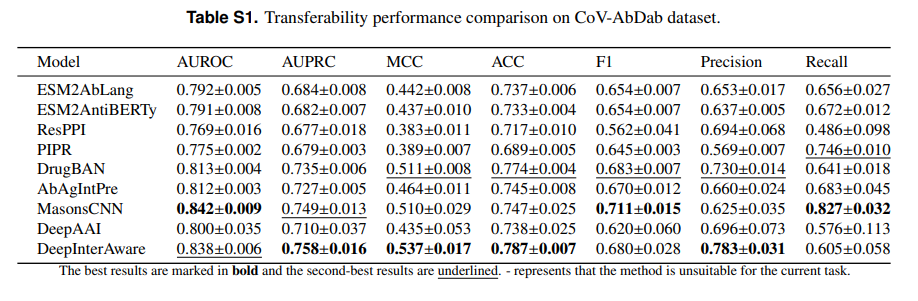
대부분의 지표에서 가장 좋은 성능을 ...(?) 흠.. 보였다고는 하는데 약간 갸우뚱 ... 괜히 Supplementary에 넣어둔건 아닌 것 같다. 넘어가자.
2.3. DeepInterAware Identifies Potential Binding Sites of Antigen–Antibody Interactions
다음으로 항원-항체 사이의 Binding Site를 identification 하는 능력에 대해서도 평가를 진행했다는데 이건 어떻게 했는지 잘 알아두면 나중에 응용하기 좋을 것 같으니 집중해서 보자.
대부분의 존재하는 AAI prediction model들은 단순히 AAI가 있는지 없는지만을 예측하는 반면, 실제로 모델을 이용하기 위해서는 AAI가 어느 부위에서 일어나는지를 해석할 수 있는 해석가능성이 매우 중요하다. 본 모델은 여러 모듈이 그런 정보를 잘 학습해서 해석 가능성을 높였다. (binding site identification)
특히 모델의 IIL (첫번째 포스팅 모델구조 참고)의 weights를 0~1 사이의 값으로 converting해서 0.5 이상으로 나오는 부위는 항원과 항체에서의 binding site로 지정하였고 나머지 비교 모델들은 그냥 모델이 지정한 default 값을 그대로 사용하였음
Sequence-based method (Honda et al.’s method[26]) as well as the structure-based methods EPI-EPMP,[27] PECAN,[28] and additionally adopted the structure-based protein binding site prediction method PesTo. (사용된 비교모델들)
Figure 2a. 2b.를 보면 recall을 통해서 우리 모델은 다른 structure based model에 뒤지지 않는 binding site prediciton preformance를 보이는 것을 확인할 수 있음.
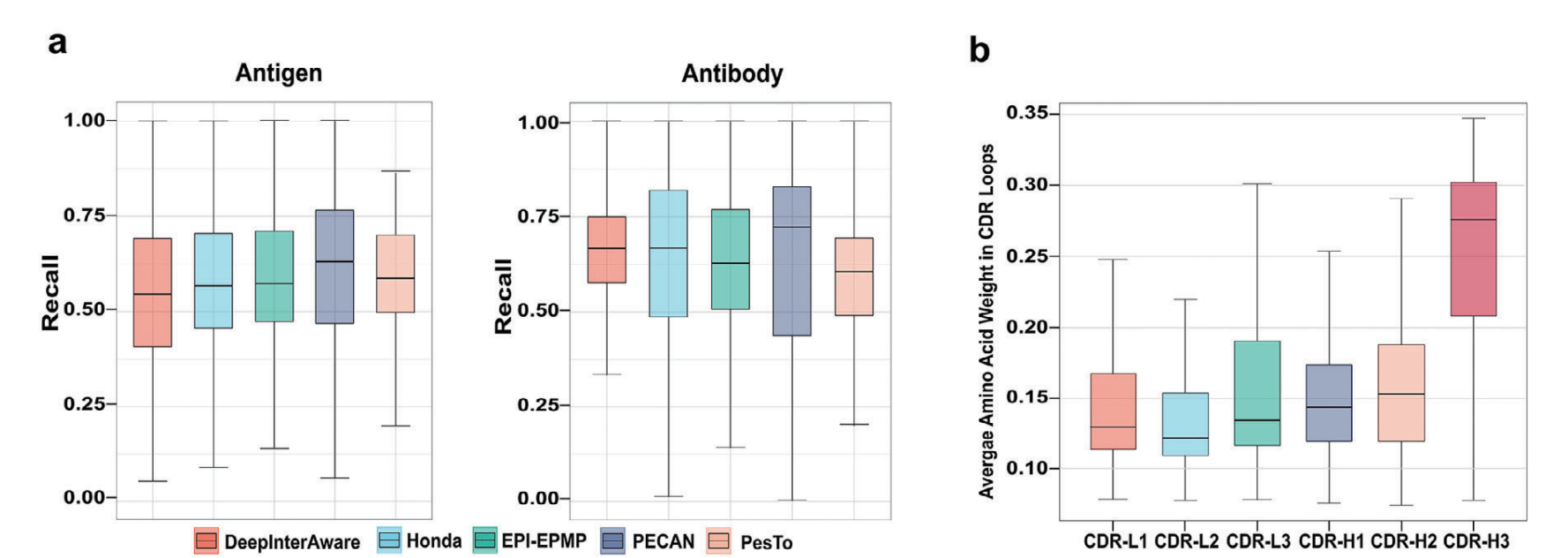
뭐 구조 데이터를 사용하는 모델들에 비해서 나쁘지 않은 수준의 recall과 CDR H3에 대한 평균적인 weight가 높은 것으로 보아서 실제로 어느 정도 binding site를 예측하는 능력이 존재한다고 볼 수는 있겠다.
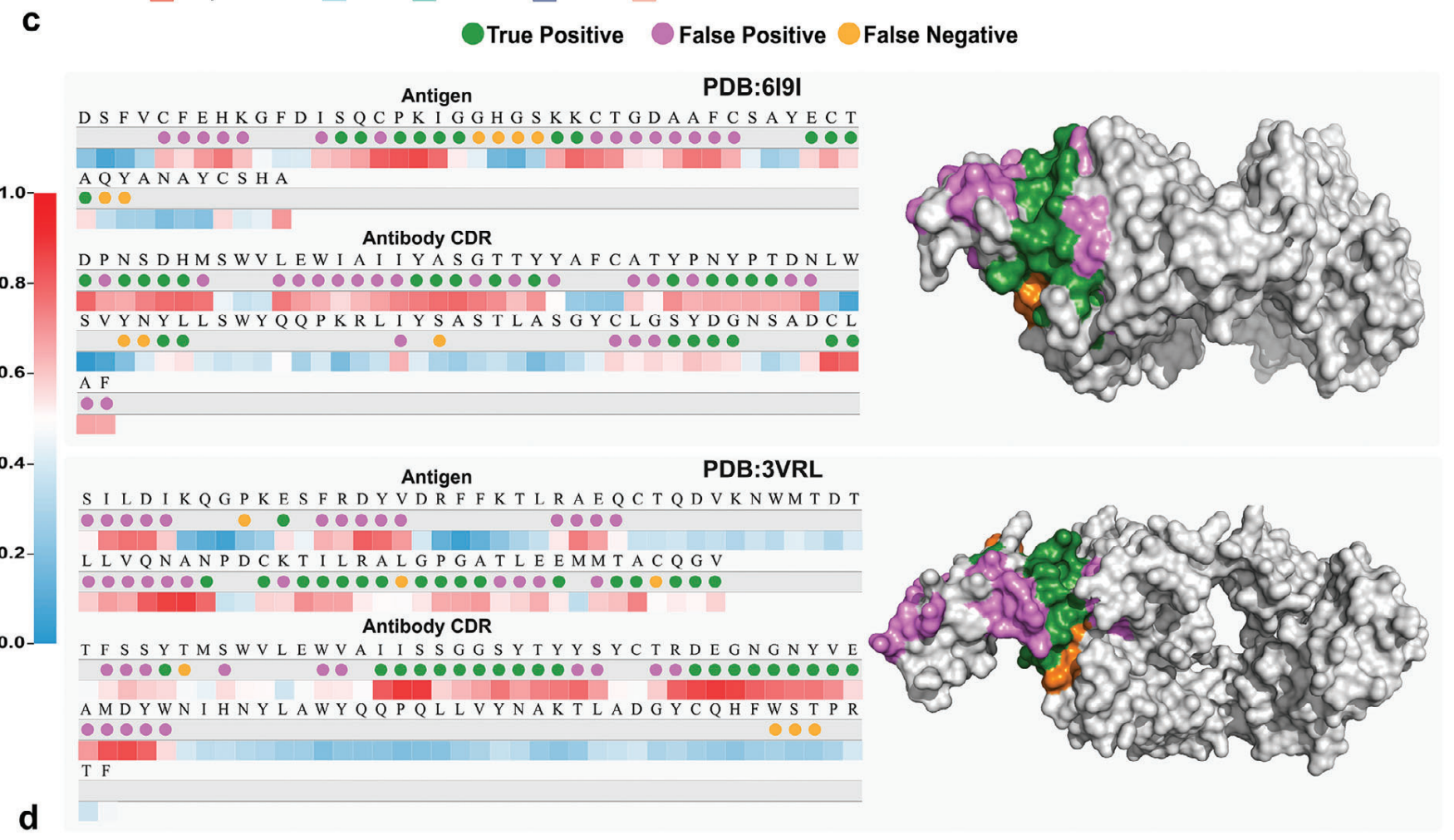
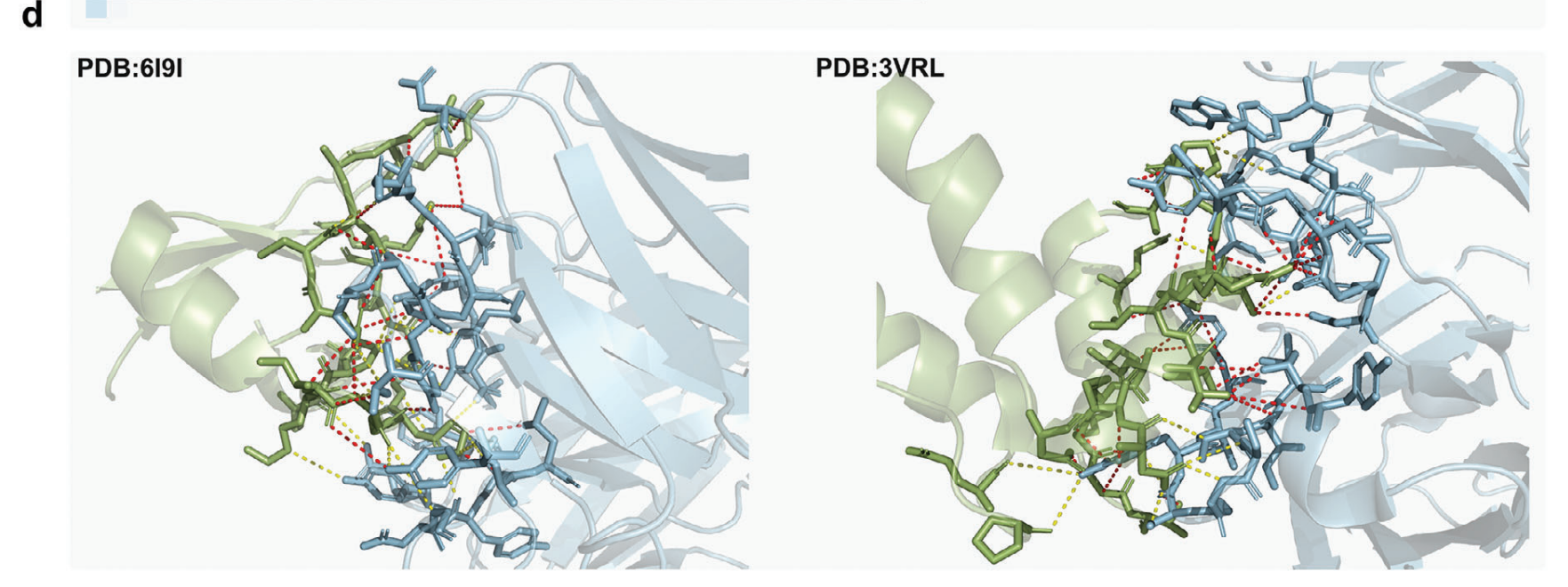
C는 실제로 어느 위치가 Attention 되어있는지도 보여주고 있음. D는 실제로 PDB파일 하나를 시각화해서 interaction을 맞춘 부분은 빨간색으로, 못맞춘 부분은 노란색으로 dash line으로 표시한 시각화 결과임. recall score로 치면 대략 56%~64% 사이의 binding pair를 실제로 identification 했다고 하여 이미지로 직관적으로 보여줘서 좋다. 이런것들은 나도 논문으로 작성할 때 참고하면 좋을 듯.
2.4. DeepInterAware Detects Mutations Within Antigens or Antibodies and Predicts the Binding Free Energy Changes
Binding Free Energy는 보통 Gibbs Free Energy라 불리며 두 물체 사이의 결합시의 안정성을 실수값으로 나타내주는 지표이다. 보통 ΔG로 표현하는 경우가 많고 값이 작은 음수값을 가질 수록 두 물체는 안정적인 결합을 하고 있다(또는 강한 결합을 한다)고 해석하면 된다.
Binding Free Energy Changes는 (ΔΔG) 말 그대로 ΔG의 변화량을 말한다. 예를들어 항체의 CDR 부위에 point mutation이 생기거나 항원의 단백질 구조가 변하면 그에 따라 항원-항체의 ΔG 값이 변하는 것은 직관적으로 자명하다. 2.4절에서는 실제로 DeepInterAware 모델이 이런 항원,항체의 서열 변화에 따른 ΔΔG 값을 얼마나 잘 예측하는지를 검증한다.
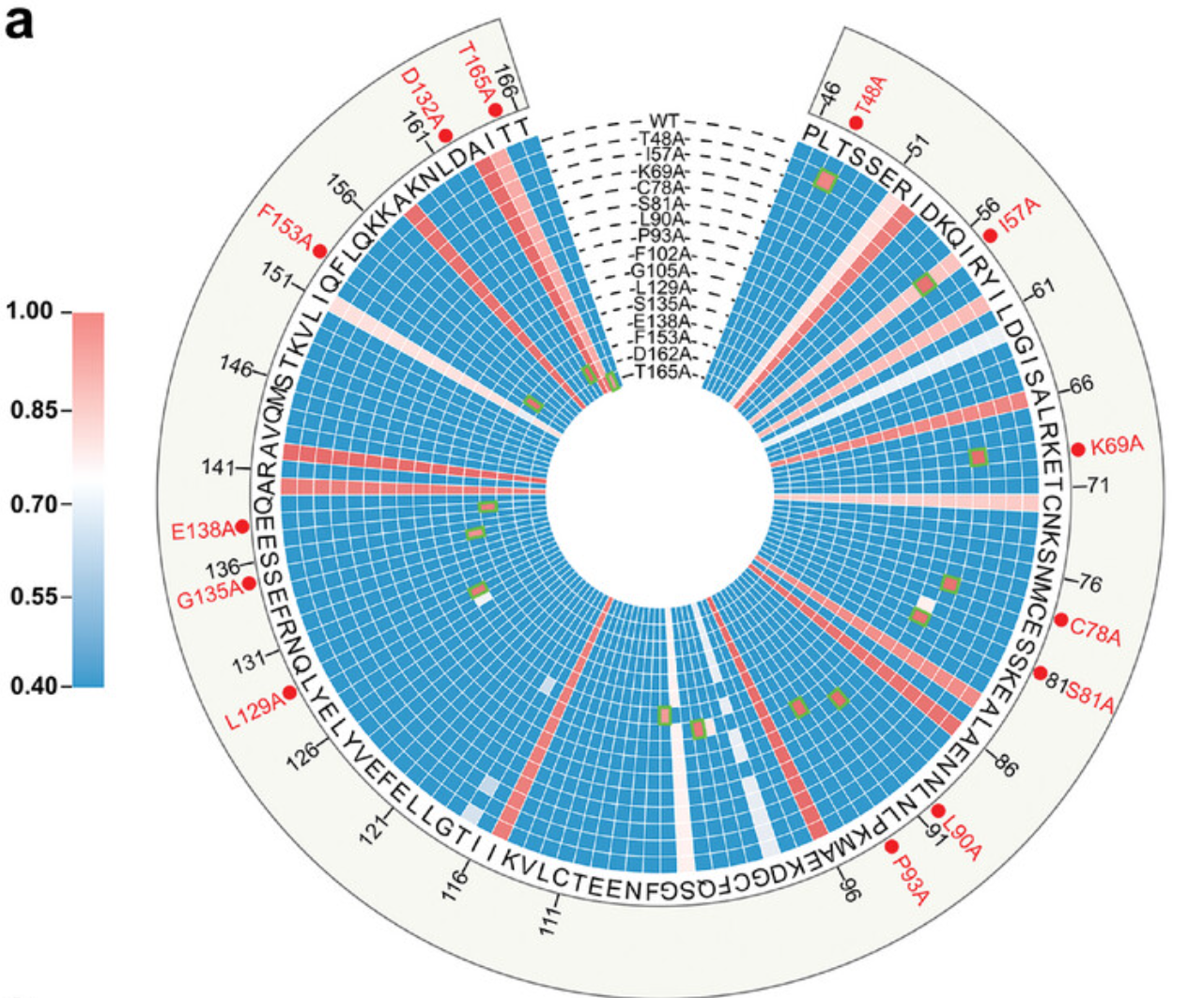

Figure a를 보면 거의 대부분의 지점에서 대부분의 attention score가 비슷하지만 특히 point mutation이 생기는 부분들에 대해서는 유독 attention socre가 높아지는 경향을 볼 수 있다. (초록색 border line으로 칠해진 주황색 네모 박스들을 주목하면 됨)
Figure b는 HC의 CDR에 point mutation을 하나씩 주면서 binding score를 예측하도록 한 값을 정리한 box plot이라고 함. 저자들은 CDR에 mutation이 생기면 항원과의 결합력이 약해질 것으로 가정하였고, 실제로 통계적으로도 그렇게 나왔다고 말하고 있음. 그런데 나는 이 실험은 인정을 못하겠음 실제로는 항원과의 affinity maturation을 위해서 CDR에 point mutation을 주고, 이 과정에서 오히려 affinity가 더 높아지는 경우도 많음. 따라서 point mutation에 의해서 perturb된 CDR이 항원에 대한 결합력이 모두 떨어지는 것만 보여지는 것은 오히려 모델이 실제 point mutation에 의한 ΔΔG를 잘 예측했다고 말하긴 어렵다고 생각함.

Figure c를 보면 AB-Bind와 SKEMPI 2.0v 데이터셋이 등장한다. 이 데이터들이야말로 ΔΔG를 수치적으로 정답값으로 주는 데이터들이라서 가장 명확하게 ΔΔG 예측 성능을 확인할 수 있는 지표임. 10-Fold CV를 진행해서 매번 나온 결과값을 저장해서 전체 데이터에 대한 plot을 찍은 것 같음.
일단 이 분야의 명확한 문제가 또 보이는게, Predicted value와 실제 value의 scale에 차이가 있다. (x, y축을 보면 됨) 이게 참 나도 DTA 모델을 만들면서 느꼈지만 Predicted model들의 대부분의 문제점은 양 극단에 위치한 값들을 잘 예측하지 못한다는 것이다. 아마도 모델의 입장에서는 대부분의 데이터 정답 값이 정규분포를 따라 평균 사이에 몰려있기 때문에 괜히 극단적인 값을 예측해서 위험부담을 질 필요가 없다는 방향으로 학습하게 되는 것 같다.
사람이 도박을 하는 이유는 무엇일까? 실패할 가능성도 높지만 성공했을 때는 그만큼 강한 보상이 돌아오기 때문이다. 그러나 도박을 하는 그 정도를 조정할 필요는 있다. 이 두가지 개념을 이용해서 이 분야의 prediction model에서 어떻게 극단에 위치한 값을 예측할 수 있도록 할지를 좀 생각해 볼 필요가 있겠다.
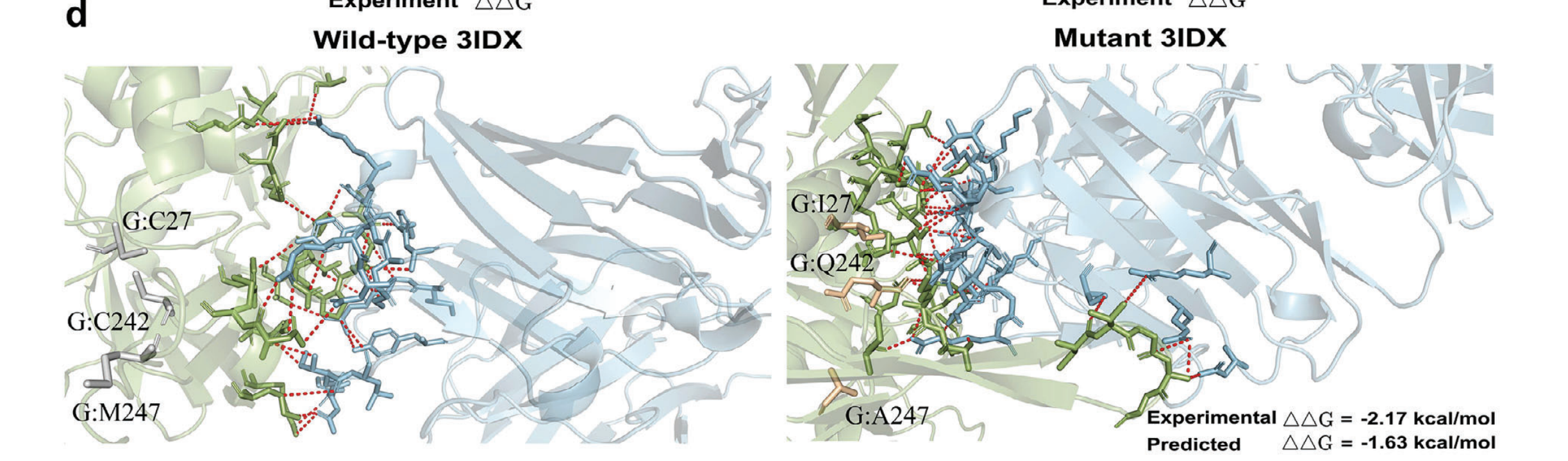
이것도 Case study로 보여주기 좋은 실험같다. Wildtype과 비교해서 Mutant를 보여주고, 실험적으로 ΔΔG값이 더 좋아진 경우를 하나 시각적으로 보여주면서 Predicted ΔΔG도 더 좋아진 것을 하나 선택해서 가장 직관적으로 와닿는 경우를 보여준다. 사실 뭐.. 체리피킹하기 너무 좋은 결과이지만 이 분야에서는 이정도 체리피킹은 어느정도 용인하는 분위기니까 나도 사용해봄직 하다.

최근에 나온 ΔΔG prediction model들을 이용해서 비교실험을 진행한 점은 매우 훌륭하다. 좋은 결과같은데 이건 왜 Supplementary에 실어놨는지 모르겠다. AttAbseq은 리뷰도 해두었으니 혹시 필요하면 읽어보시길, 근데 썩 좋은 구성의 논문은 아니었음 https://biomadscientist.tistory.com/184
2.5. DeepInterAware Screens the Potential Antibodies Binding to HER2 Target
마지막으로 DeepInterAware를 이용해서 HER2 단백질과 interaction 할 것으로 생각되는 후보 antibody를 screening하는 실험을 진행했음. 이것도 Case Study로 넣기 너무너무 좋은 실험이라 아주 훌륭한 Case Study. 좋은 논문은 괜히 좋은게 아니다.
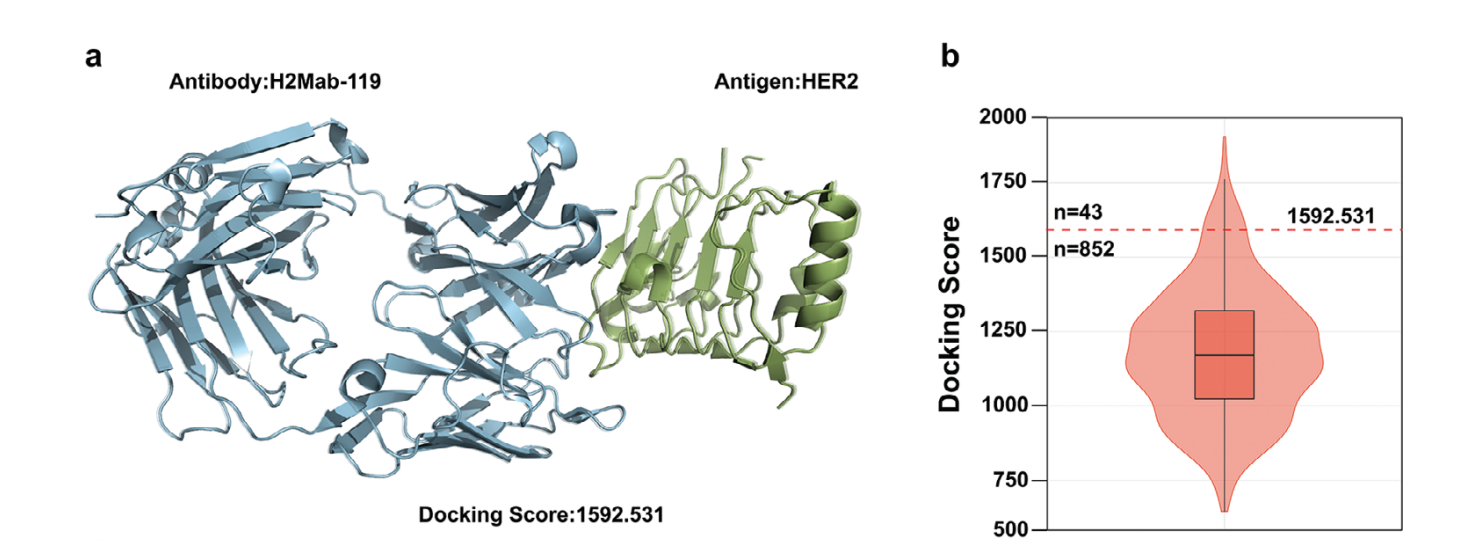
Figure a는 실제로 HER2와 Binding하는 것으로 조사된 항체와의 결합 구조체임. 이것을 따로 떼어서 항원과 항체로 분리한 다음 ZDock을 이용해서 다시 결합시켜서 Docking Score를 계산하고 이 값을 Ground Truth Threshold로 사용함. 이 값보다 더 좋은 Docking socre를 가지는 항체를 screening 하는 것을 목표로 함.
Figure b에 실제로 screening한 결과의 바이올린 플롯을 볼 수 있음.
스크리닝 과정
- HER2–H2Mab-119 복합체를 항원과 항체로 분리.
- ZDOCK을 이용해 이 복합체의 global docking score를 계산 → baseline으로 사용.
- DeepInterAware를 사용해 SAbDab 데이터베이스 내 11,683개 항체를 HER2에 대해 스코어링.
- 예측 binding score가 0.95 이상인 895개 항체를 선별.
- 이들 후보군을 ZDOCK으로 다시 HER2에 도킹해 검증.
결과
- 총 43개 항체 후보가 H2Mab-119보다 더 높은 docking score를 기록.
- 그 중 Fab-bound IDE (PDB: 4M1C) 가 가장 높은 점수.
- 또 다른 후보인 NabFab (PDB: 7RTH) 은 HER2에 H2Mab-119와 매우 유사한 부위에서 결합하며, 결합 인터페이스 아미노산의 85%가 겹침.
- 따라서 NabFab은 HER2를 표적으로 하는 유망한 치료 항체 후보로 제시됨.
총평으로, 모델도 엄청나게 어렵지 않으면서도 좋은 성능을 보였고, 상당히 다양한 실험 검증과 함께 많은 비교실험을 철저하게 진행한 점 등, 사소한 몇몇 생물학적 검증과는 맞지 않는 개념들 말고는 논문의 구성이 아주 탄탄하다. 솔직히 여기서 in vitro 실험까지만 있었으면 Nature Machine Intelligence도 가능했을 것 같다. 모름지기 응용분야의 논문은 이렇게 독자들이 읽었을 때 의문스러운 점이 없이 동화책 읽듯이 술술 읽혀야 한다고 생각한다. 오랜만에 아주 좋은 논문을 읽어서 기분이 좋다. 앞으로 논문을 작성할 때도 이 논문의 구성을 따르면 도움이 많이 될 것 같다.
'Paper > Bioinformatics' 카테고리의 다른 글
| DeepInterAware 논문 리뷰 (1) 모델 구조 (1) | 2025.08.25 |
|---|---|
| AttABseq 논문 리뷰 (4) | 2025.08.22 |
| AbSet 논문리뷰 (3) | 2025.08.19 |
| [2022 NIPS] DIffAb 논문리뷰 (1) (0) | 2025.03.27 |
| HyperAttentionDTI 논문 리뷰 (4) | 2023.11.24 |
- Total
- Today
- Yesterday
- manim library
- marginal likelihood
- 이왜안
- 기계학습
- manim
- antigen antibody interaction prediction
- dataloader
- 오일석기계학습
- 3b1b
- eigenvector
- 선형대수
- 인공지능
- manimtutorial
- 파이썬
- 항원항체결합예측모델
- eigenvalue
- 논문리뷰
- 3B1B따라잡기
- nanobody
- MatrixAlgebra
- ai신약개발
- MLE
- 최대우도추정
- 백준
- 나노바디
- Matrix algebra
- Manimlibrary
- MorganCircularfingerprint
- 베이즈정리
- elementry matrix
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |